the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Feb 2025
| 05 Feb 2025
The effect of carbonate mineral additions on biogeochemical conditions in surface sediments and benthic–pelagic exchange fluxes
Kadir Biçe
Tristen Myers Stewart
George G. Waldbusser
Coastal sediments are hotspots of biogeochemical processes that are impacting subsurface and overlying water conditions. Fluid composition in sediments is altered through the mineralization of organic matter which, under oxic conditions, further lowers both pH and the carbonate saturation state. As a potential mitigation strategy for this sediment acidification, we explored the effects of mineral additions to coastal sediments. We experimentally quantified carbonate mineral dissolution kinetics of carbonate shells suitable for field application and then integrated these data into a reactive transport model that represents early diagenetic cycling of C, O, N, S, and Fe and traces total alkalinity, pH, and saturation state of CaCO3. Model simulations were carried out to delineate the impact of mineral type and amount added, porewater mixing, and organic matter mineralization rates on sediment alkalinity and its flux to the overlying water. Model results showed that the added minerals undergo initial rapid dissolution and generate saturated conditions demonstrating the potential of alkalinity enhancement in mitigating surface sediment acidification. Aragonite dissolution led to higher total alkalinity concentrations than calcite. Simulations of carbonate mineral additions to sediment environments with low rates of organic matter mineralization exhibited a substantial increase in mineral saturation state compared to sediments with high CO2 production rates, highlighting the environment-specific extent of the effect of mineral addition. Our work indicates that carbonate additions have the potential to effectively buffer surficial sediments over multiple years, yielding biogeochemical conditions that counteract the detrimental effect of low-pH sediment conditions on larval recruitment and potentially increase benthic alkalinity fluxes to support marine carbon dioxide removal (mCDR) in the overlying water.
- Article
(1664 KB) - Full-text XML
-
Supplement
(657 KB) - BibTeX
- EndNote




In coastal systems, ocean–freshwater mixing, terrestrial inputs, and active biogeochemical transformations (Cai et al., 2017; Soetaert et al., 2007) lead to natural fluctuations in pH that are large and decoupled from the decrease in pH observed in the open ocean due to ocean acidification (Hofmann et al., 2011). Especially in shallow-water environments, sediments play a significant role as benthic fluxes can constitute a substantial alkalinity source to the overlying water, increasing its buffering capacity (Rassmann et al., 2016; Krumins et al., 2013; Cyronak et al., 2013b). Sediments receive organic matter (OM) from the overlying water, which fuels the dissolution of carbonate minerals and early diagenetic processes. Aerobic respiration and other processes consuming dissolved O2 can lead to steep pH decreases within the oxygenated sediment and the interaction of OM mineralization, and mineral dissolution determines the balance of the porewater carbonate system (Burdige et al., 2010; Jourabchi et al., 2005). Mineral dissolution and early diagenesis are connected through the changes in carbonate system, which impacts mineral saturation states and hence the rate of dissolution (e.g., Morse et al., 2007). Apart from its impact on mineral dissolution, OM mineralization is also a significant contributor to alkalinity generation under suboxic and anoxic conditions (Hu and Cai, 2011a). These early diagenetic processes determine the composition of the porewaters, which affect benthic calcifying organisms; furthermore, they can affect the biogeochemical conditions in the overlying water through benthic fluxes releasing nutrients and alkalinity.
In surface sediments, porewater pH is generally lower than in the overlying water, in particular when sediment O2 uptake is elevated due to burrowing and irrigation activities by benthic macrofauna (e.g., Aller, 1982). This exposes benthic organisms that live in the upper layers of the sediment to more corrosive conditions (Green et al., 2013; Waldbusser and Salisbury, 2014). Combined with abundant OM, enhanced supply of O2 into the sediment also leads to elevated fluxes of alkalinity and dissolved inorganic carbon (DIC) to the water column (Mucci et al., 2000; Burdige, 2007).
In this study, we investigate the effects of the addition of carbonate minerals in the coastal sediments focusing on benthic alkalinity generation and release in a shallow-water environment. Such ocean alkalinity enhancement (OAE) aims to buffer increased dissolved CO2 in the ocean by dissolving carbonate or silicate minerals, producing alkalinity. OAE is mainly studied for its effect on ocean–atmosphere CO2 exchange, the impact on organisms (Renforth and Henderson, 2017), and its potential to mitigate ocean acidification and lower atmospheric CO2 concentrations (marine carbon dioxide removal; mCDR). Previous studies have shown that increasing ocean alkalinity through enhanced weathering has the potential to reverse global ocean acidification in an intermediate CO2 emission scenario (RCP 4.5) and reduce its impacts if emissions were to continue to rise throughout the 21st century (RCP 8.5) (Taylor et al., 2016). To date, OAE implementations mainly have targeted the open ocean and have been aimed at identifying the potential of scaling up the mCDR efforts to a global scale (Fennel et al., 2023). Recent studies also focused on the impact of OAE on marine ecosystems such as fish and plankton communities (Goldenberg et al., 2024; Paul et al., 2024; Ferderer et al., 2022). Only a few studies have focused on coastal OAE and even fewer on sedimentary applications (Green et al., 2009, 2013; Hangx and Spiers, 2009; Fuhr et al., 2024), where the effectiveness of OAE mainly depends on the dissolution rate of the mineral which is controlled by the mineral and the chemistry of the water and physical properties of the environment (Hartmann et al., 2013; Montserrat et al., 2017). One benefit of implementing OAE in coastal sediments is the generally higher rate of organic matter remineralization. This elevated CO2 generation aids mineral dissolution, given most overlying marine waters are typically super-saturated with respect to carbonate minerals. Here, we simulate this effect of adding carbonate minerals to the sediment using a reactive transport model. We use measurements of carbonate mineral dissolution rates to assess the extent and duration of buffering in surficial sediments and the impact on benthic alkalinity fluxes. Building on the previous work by Krumins et al. (2013) that explored the interaction of carbonate chemistry with early diagenesis in coastal sediments, our work identifies factors that determine the impact of sediment alkalinization on the spatial and temporal distribution of buffering by exploring the effect of mineral type, amount, organic matter remineralization rate, and bioturbation.
2.1 Dissolution experiments
Dissolution rates for the minerals used in this model were derived from lab-based experiments detailed in Myers (2022). We provide pertinent details below. Two biomineral calcium carbonates – calcitic oyster shells of Crassostrea gigas (below referred to as “biocalcite”) and aragonitic soft-shell clam shells of Mya arenaria (“bioaragonite”) – were collected from Oregon Oyster Farm in Newport, Oregon, USA, and from Broad Cove in Yarmouth, Maine, USA, respectively. The biomineral samples were briefly cleaned, dried, crushed, and sieved through a 2000 µm sieve and retained on 710 µm, for an approximately median grain size of 1 mm. We measured the mineral dissolution rate across four saturation states experimentally; Ωcalcite=0.25 (low), 0.45 (middle), 0.85 (high), and 1.03 (saturated); carbonate chemistry conditions are detailed in Table S1 in the Supplement. A flow-through, feedback-controlled with pH monitoring and a CO2 injection seawater system similar to Waldbusser et al. (2011), was used for manipulations at the Hatfield Marine Science Center (HMSC), Newport, OR, USA (additional details in Myers, 2022). pH measurements from each manipulation tank were compared against a benchtop meter daily during the experiment. pH was additionally compared against a reference sample in which pCO2 and TCO2, in situ temperature, and salinity were measured to calculate pH on the NBS scale. pH (all tanks) and temperature (low and pretreatment tanks) data were recorded every 10 min. The coefficient of determination (R2) between measured benchtop pH data and the pH data calculated from pCO2 and TCO2 measurements for header tanks in all experiments was consistently greater than 0.9 across all experiments (Fig. S2 in the Supplement). Plug-flow style dissolution chambers were 2.5 cm i.d., 12.7 cm long, schedule 40 PVC pipe, with 150 µm Nitex mesh on either end of the upright chambers. Approximately 50 (±0.1) g of biomineral was used in each chamber; water was initially added slowly and agitated to remove air bubbles prior to initiating flow. Flow rates were controlled to 17 (±1.2) mL min−1 and experiments were run for 3–4 d to verify consistent alkalinity concentrations, as observed in trials conducted to optimize the experiment (Myers, 2022). Using the inlet and outlet measured alkalinity concentrations from an individual chamber (mol alkalinity L−1), the measured flow rate (L d−1), and the known amount of sample added (gmineral), the change in alkalinity in mol alkalinity d−1 was recorded as Eq. (1):
The early diagenetic model (see below) uses a rate law in which the volumetric dissolution rate Rd (in molmineral m d−1) depends on the concentration of the mineral (molmineral m), the saturation state Ω (= ion concentration product/solubility Ksp), and the dissolution rate constant kd (d−1) (Jourabchi et al., 2008):
In the experiment, the dissolving mineral is the entire solid phase so that gmineral=gsolid phase. Hence, the mineral concentration in mol is the molecular weight of the mineral (MWmineral), and kd can be calculated from the measured Δ as
where dTA/dR is the mineral to alkalinity stoichiometric ratio in the dissolution reaction (+2 molalkalinity / molmineral; Table 3) and n is the rate order.
2.2 Carbonate chemistry measurements
2.2.1 Alkalinity
Open-cell alkalinity titrations were conducted over the course of the experiment, utilizing a two-point titration after Edmond and Gieskes (1970), as modified by Waldbusser et al. (2011). A micrometer burette was used with a micro pH probe with precisions of 0.002 mL and ±0.02, respectively. The probe was calibrated with NBS calibration standards (pH =4, 7, and 10) daily before being used to take measurements. As outlined in Waldbusser et al. (2011), Baker Analyzed Reagent grade 0.0995–0.1005 normal HCl acid was used as the titrant with reagent grade NaCl to a final molar concentration of 0.7 to minimize gap junction potentials during titrations. Analytical precision and accuracy were compared against triplicate titrations and Dickson CRM batch 142, respectively, and found to have an average coefficient of variance of 0.07 % and typically ±16 µmol or less, for precision and accuracy.
2.2.2 Dissolved inorganic carbon, pCO2, and carbonate calculations
pCO2 and TCO2 samples were collected in clean amber glass ∼350 mL bottles preserved with 30 µL of saturated HgCl2 and sealed with polyurethane-lined crimp-sealed metal caps. Analysis via the Burkalator was carried out to measure pCO2 and TCO2 via near-infrared detection following the procedure of Bandstra et al. (2006), modified for discrete samples as in Hales et al. (2005), Barton et al. (2012), and Hales et al. (2017). Liquid and gas standards were employed to ensure the accuracy of pCO2 and TCO2 measurements. The system can resolve the accuracy and precision of TCO2 concentrations within ±0.02 % and 2 % for pCO2 (Hales et al., 2017). Carbonate chemistry parameters were calculated using dissociation constants from Millero (2010). Salinity was measured on the sample bottles following pCO2 and TCO2 measurements via a salinometer (8400B Autosal Laboratory Salinometer; Guildline Instruments, Sorrento, FL, USA). Calculated carbonate chemistry variables presented in association with the mineral dissolution experiments were computed using CO2calc and the K1 and K2 carbonic acid dissociation constants from Lueker et al. (2000), constants from Dickson (1990), sulfate constants from Morris and Riley (1966), fluorine constants from Riley (1965), and boron constants from Uppström (1974).
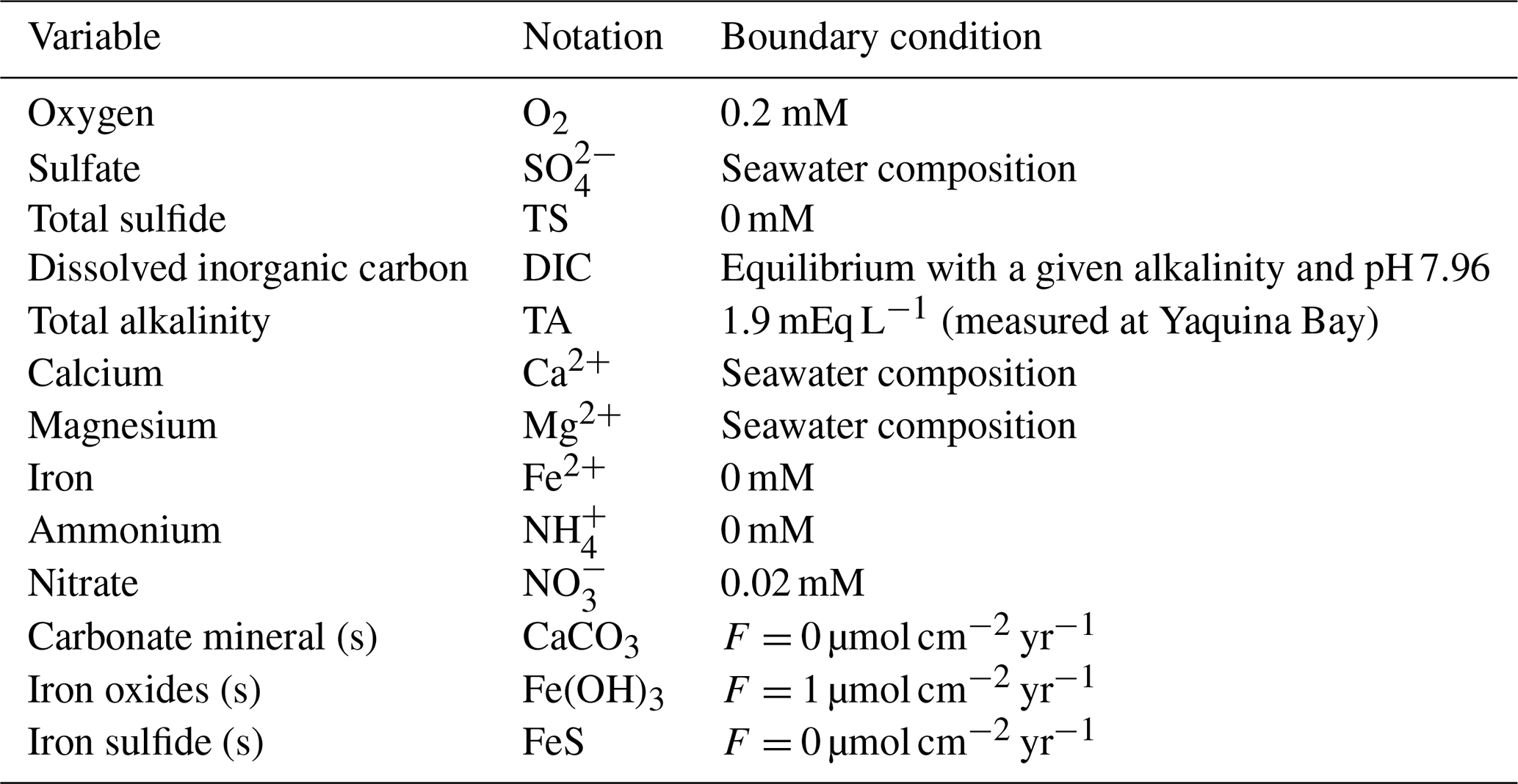
Table 1State variables and their upper boundary conditions. Seawater compositions are computed with AquaEnv (Hofmann et al., 2010) for the pressure, temperature, and salinity given in Table 2.
2.3 Reactive transport modeling
A one-dimensional diagenetic model was developed to simulate the distribution of chemical species with depth in the sediment over time. The model domain extended 20 cm deep into the sediment discretized into 50 intervals with a linearly increasing grid size of 1 mm at the top and 1 cm at the bottom. The model included 13 state variables, representing chemicals involved in the main early diagenetic reactions and mineral dissolution/precipitation (Table 1). At the upper boundary, the concentrations of solutes and fluxes of solids were imposed, while at the bottom boundary, no gradient conditions were imposed for all chemicals.
The spatiotemporal evolution of the concentrations was described by an advection–diffusion equation with the addition of bioturbation and bioirrigation and the effect of reactions. Bioturbation was treated as a diffusive process (Boudreau, 1997), and bioirrigation was described as a non-local exchange between porewater at depth and the overlying water (Boudreau, 1984). The governing equations were
where t is time, x is depth in sediment, ϕ is porosity, Ci (Cj) is the concentration of the solute i (in mol per volume pore fluid; solid j in mol per volume solid phase), u is the burial velocity for solutes (v for solids), Di is the diffusion coefficient of the solute i, Db is the depth bioturbation coefficient, Rr is the rate of production/consumption through reaction r (in mass per total volume and time), s is the stoichiometric coefficient for solute i or solid j in reaction r, and Bi represents bioirrigation for solute i.
Porosity is constant with time but depth-dependent and exponentially decreases with depth, reflecting steady-state sediment compaction (Eq. 6):
where ϕ∞ and ϕ0 are the porosities at infinite depth and at the top of the sediment, respectively, and γ is the e-folding distance for the porosity. Burial velocities were corrected for compaction (Berner, 1980):
Diffusion coefficients were calculated using the R package marelac after Boudreau (1997) at a given salinity, temperature, and pressure (Soetaert et al., 2010) and corrected for tortuosity (Boudreau, 1996) so that
where Daq is the molecular diffusion coefficient of solute i in solution. The bioturbation coefficient decreases exponentially with depth below the mixed layer:
where xm is the depth of the mixed layer and Db0 is the bioturbation coefficient at the sediment–water interface (SWI), calculated as a function of water depth (Middelburg et al., 1997).
Bioirrigation is defined as an exchange between the surface layer and deeper layers of the sediment, which was constrained by a bioirrigation coefficient exponentially decreasing with depth:
where α0 is the bioirrigation coefficient at the SWI, ε is the e-folding distance for bioirrigation, and Ci,x is the concentration of solute i at depth x.
Table 3Reactions included in the model. Benthic primary production is assumed to be negligible, and organic matter has Redfield stoichiometry (a=1, , ). Alkalinity production or consumption in each reaction is determined following Wolf-Gladrow et al. (2007). The total mineralization rate is defined as after Wang and Van Cappellen (1996), where γom is e-folding distance for OM mineralization.
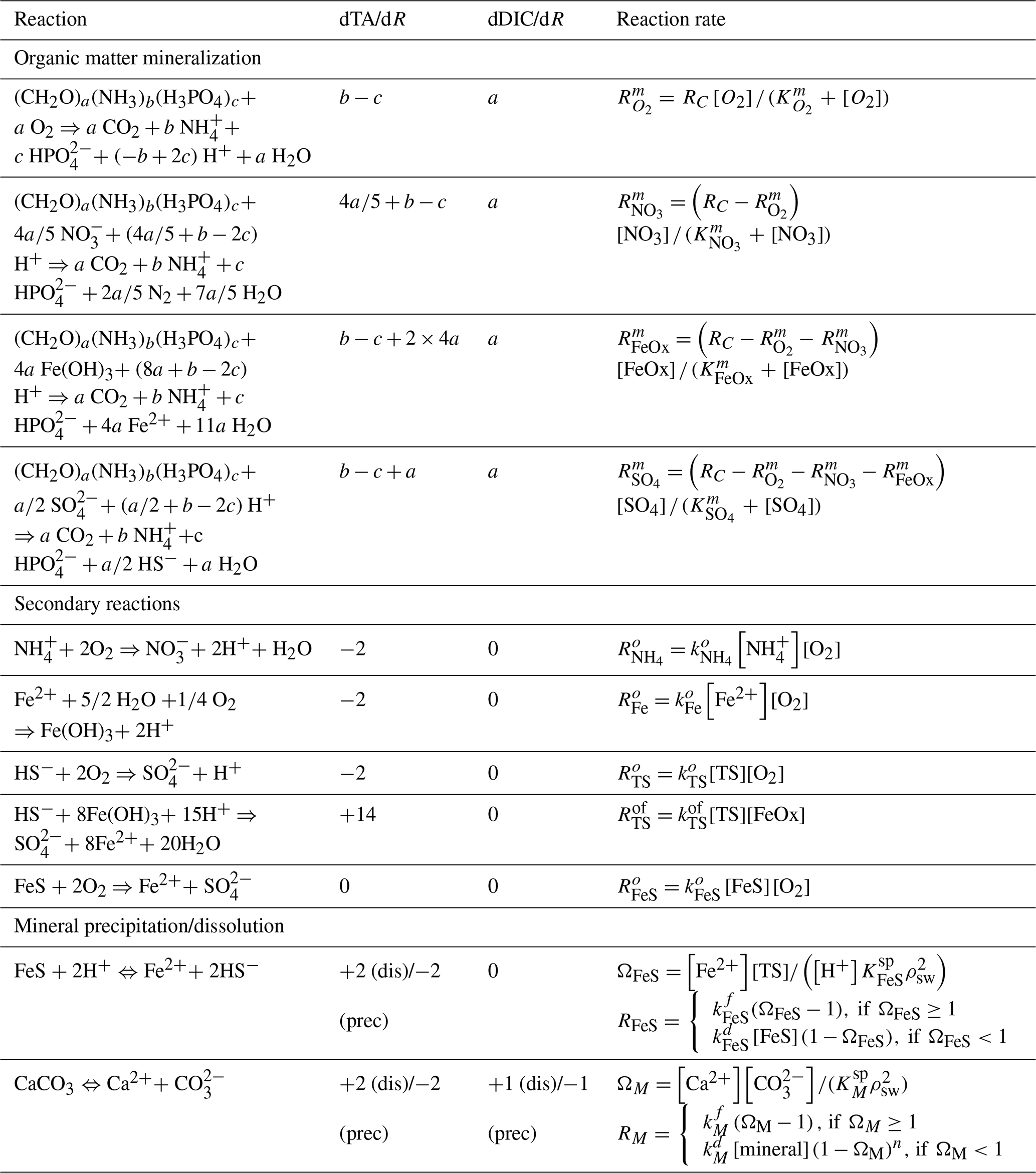
Reactions considered in the model include organic matter mineralization with aerobic and anaerobic pathways, reoxidation of reduced species, and mineral formation/dissolution reactions (Table 3). Each reaction has an impact on the total alkalinity (TA), which was taken from previous studies (Wolf-Gladrow et al., 2007; Dickson, 1981).
Organic matter is mineralized using O2, NO, iron (oxyhydr)oxides (FeOx), and SO as terminal electron acceptors and modeled with Monod kinetics. The OM mineralization rate () was adjusted to yield a TA flux of ∼1200 µmol cm−2 yr−1. This value was chosen to match open-core top incubations measured in Yaquina Bay (Myers, 2022), with the resulting value being comparable to earlier measurements by D'Andrea and DeWitt (2009) (∼200–460 µmol C cm−3 yr−1) and higher than values reported for deeper coastal environments (Wang and Van Cappellen, 1996; Krumins et al., 2013). Reactions considered further include nitrification, iron oxidation, sulfide oxidation with O2 and FeOx, and iron sulfide oxidation, as well as the formation/dissolution of CaCO3 minerals and FeS (Table 3). Following Rooze et al. (2020), at each time step, DIC is speciated to calculate CO concentration and pH from DIC and alkalinity, assuming seawater concentrations of borate and negligible contributions of phosphate, silicic acid, nitrite and hydrogen fluoride, and other acids or bases.
Table 4Reaction constants. Carbonate dissolution rate constants determined experimentally in this study are described in the results.
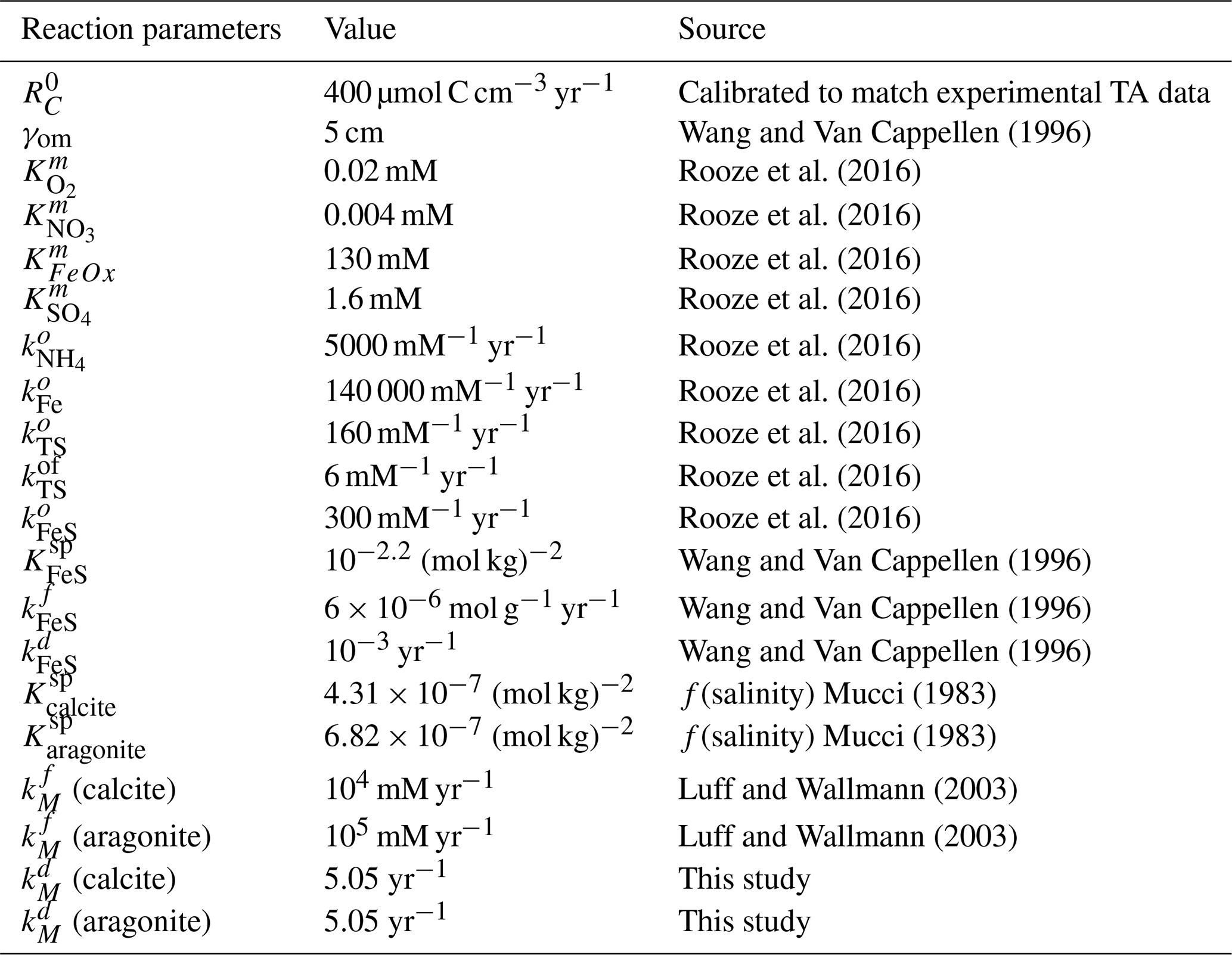
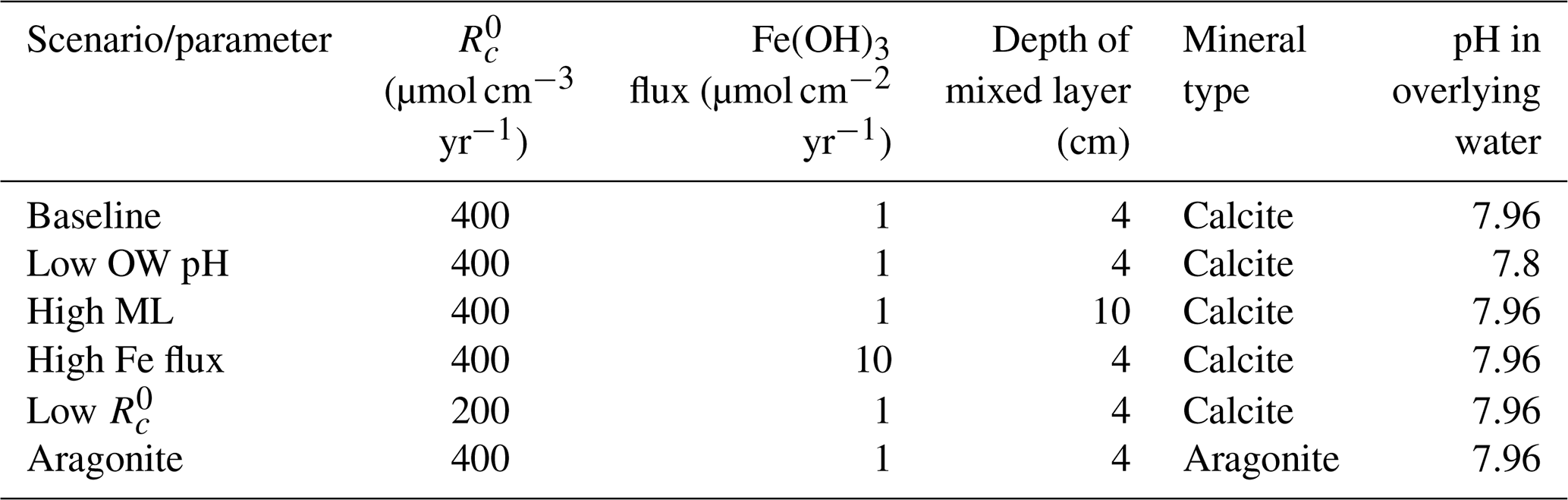
Table 5Sensitivity analyses. Rows represent the scenario tested. Columns show the parameters changed. OW – overlying water; ML – mixed-layer depth.
2.3.1 Application
To establish initial conditions, each simulation was first run to steady state with a zero-deposition flux of calcium carbonate. After that, 8 wt % or 16 wt % (weight of mineral/total weight of the sediment) CaCO3 was added to the top 2 cm of sediment, representing the sudden, one-time addition of the buffering carbonate minerals. To assess the magnitude and temporal evolution of the impact of the alkalinization experiment on sediment biogeochemistry and benthic fluxes, transient simulations using these new initial conditions were then carried out, covering periods of up to 50 years.
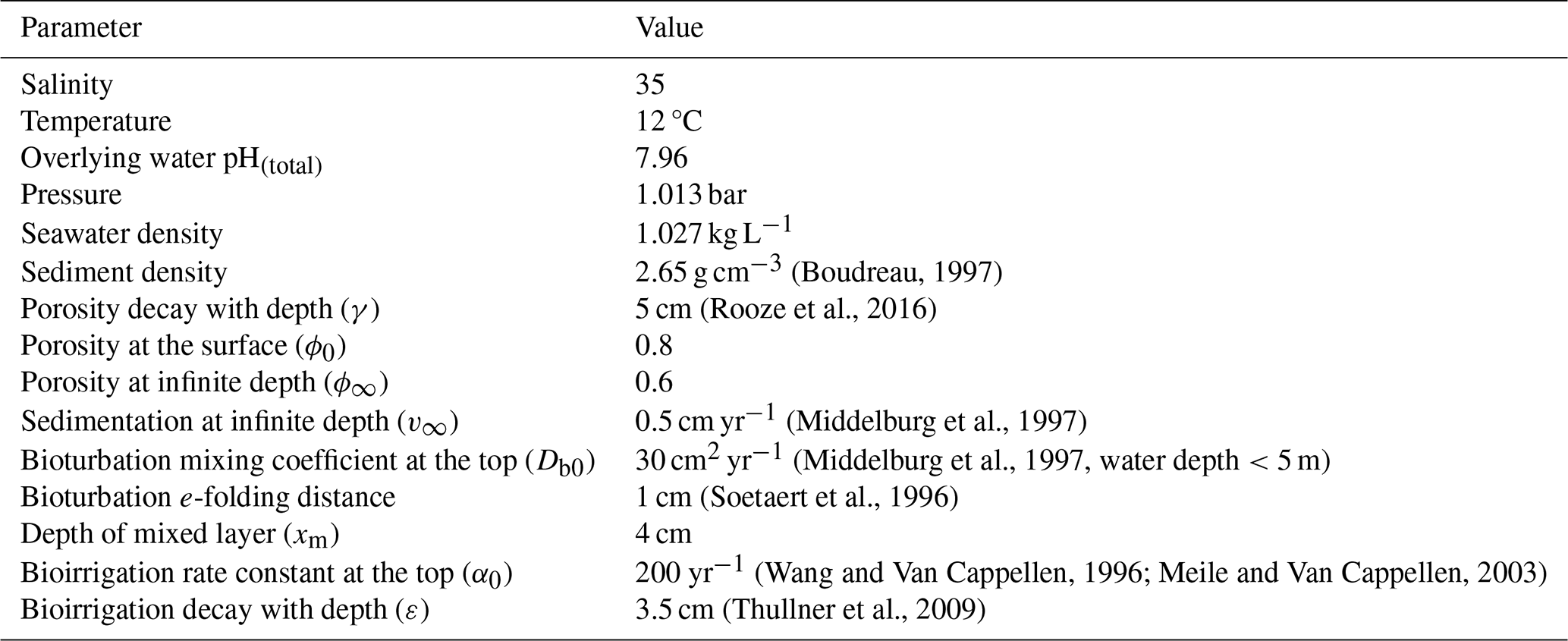
We tested the impact of several environmental factors on the buffering effect of carbonate mineral additions to surficial sediments in comparison to the baseline addition scenario (Table 5). Specifically, we lowered the overlying water pH from 7.96 to 7.8, representing the impact of ocean acidification. The depth of the bioturbated layer affects the distribution of minerals added, which may impact the effectiveness of the extent of buffering; it was varied from the relatively shallow mixing depth of 4 cm in the baseline implementation to the global average depth of bioturbation of 10 cm (Boudreau, 1997). We also varied the deposition flux of iron (oxy)hydroxides from 1 to 10 µmol cm2 yr−1 to assess differences between sites that vary in the extent of coupling between Fe, S, and C cycles. And as the rate of organic matter mineralization has a major impact on sediment redox conditions and porewater composition, was varied from the baseline value of 400 to 200 µmol cm−3 yr−1 to represent environments with different O2 penetration depths. Finally, simulations were carried out to assess the impact of the type of mineral in the system (calcite vs. aragonite). The spinup simulations were specific to each model run and as such represent the environmental conditions (e.g., high vs. low rate of mineralization or a setting in which either aragonite or calcite are the only carbonate mineral considered). Simulations were performed in R using the ReacTran package (Soetaert and Meysman, 2012).
2.3.2 CO2 drawdown
We estimated the potential impact of enhanced sediment TA fluxes on atmospheric CO2 uptake in an enclosed bay using a simple box model. Water exchange is due to the tides, and riverine impact was assumed negligible, reflecting for example the strong coupling of Yaquina Bay with the coastal ocean (Brown and Ozretich, 2009).
To estimate the order of magnitude of the impact of mineral additions on CO2 uptake, we used a simple mass balance approach similar to Brenner et al. (2016), which focuses on the ratio of DIC over TA as the controlling factor of CO2 uptake (Egleston et al., 2010). We calculated the mass balance for TA and DIC prior to and 2 years after the mineral addition and attributed the difference in atmospheric CO2 exchange to the mineral addition. As described in detail in the Supplement, we first estimated tidal water exchange from water level measurements. The characteristics of the inflowing marine (alkalinity =2.2 mmol L−1, pH =8.0) and outflowing bay water (alkalinity =1.9 mmol L−1, pH =7.96) were chosen to be representative of a coastal setting such as Yaquina Bay and were informed by unpublished temperature, salinity, and pH data (George Waldbusser, personal communication, 2024). Combined with our modeled benthic exchange fluxes, this allowed us to estimate the alkalinity production in the water column (RTA; Eq. 12). At steady state, the alkalinity balance is given by
where is the tidal influx of TA as a function of water exchange rate and oceanic TA (), is the tidal outflux of TA from the bay to the ocean as a function of tidal velocity and TA of the study site (), is the benthic flux computed by the reactive transport model, and RTA is the areal net rate of water column TA production/consumption resulting from primary productivity and respiration.
Next, we used the alkalinity and pH to calculate the DIC and converted the net alkalinity production rate into net DIC production in the water column, assuming Redfield stoichiometry. This finally allowed us to estimate the CO2 exchange flux () necessary to close the DIC mass balance for benthic fluxes with and without mineral additions to quantify its effect on ocean CO2 uptake.
The mass balance for DIC is given by
where is the tide-driven influx of DIC (), is the outflux of DIC (), is the benthic flux of DIC, is the atmospheric CO2 uptake (negative values indicate outgassing), and RDIC is the net areal reaction rate producing/consuming DIC.
3.1 Mineral dissolution rates
Dissolution rates of minerals were highest under highly undersaturated conditions (Fig. 1). Dissolution rates are approximately proportional to the degree of saturation. Thus, the dissolution rate constant kd for each mineral was estimated using Eqs. (1) and (2) with a dissolution order (n) of 1. However, we acknowledge the large variability in mineral dissolution dynamics (especially n) in previous studies (see, e.g., Adkins et al., 2021; Subhas et al., 2018), which is a critical factor for the evolution of the dissolution rate over time. Our results are in line with previous data synthesis studies focused on mineral dissolution (e.g., Morse et al., 2007; Jourabchi et al., 2005, 2008). The kinetic constant for aragonite dissolution was previously reported as 5 yr−1 (Luff and Wallmann, 2003), which was comparable to the 5.2 yr−1 derived from our measurements. For n=1, Jourabchi et al. (2005) reported a dissolution rate constant of 3.65 yr−1, while our estimation for biocalcite was 5 yr−1. Measured dissolution rates did not show significant differences between two mineral types (t test with p value >0.05). Therefore, a kinetic constant for both minerals was calculated as 5.05 yr−1.
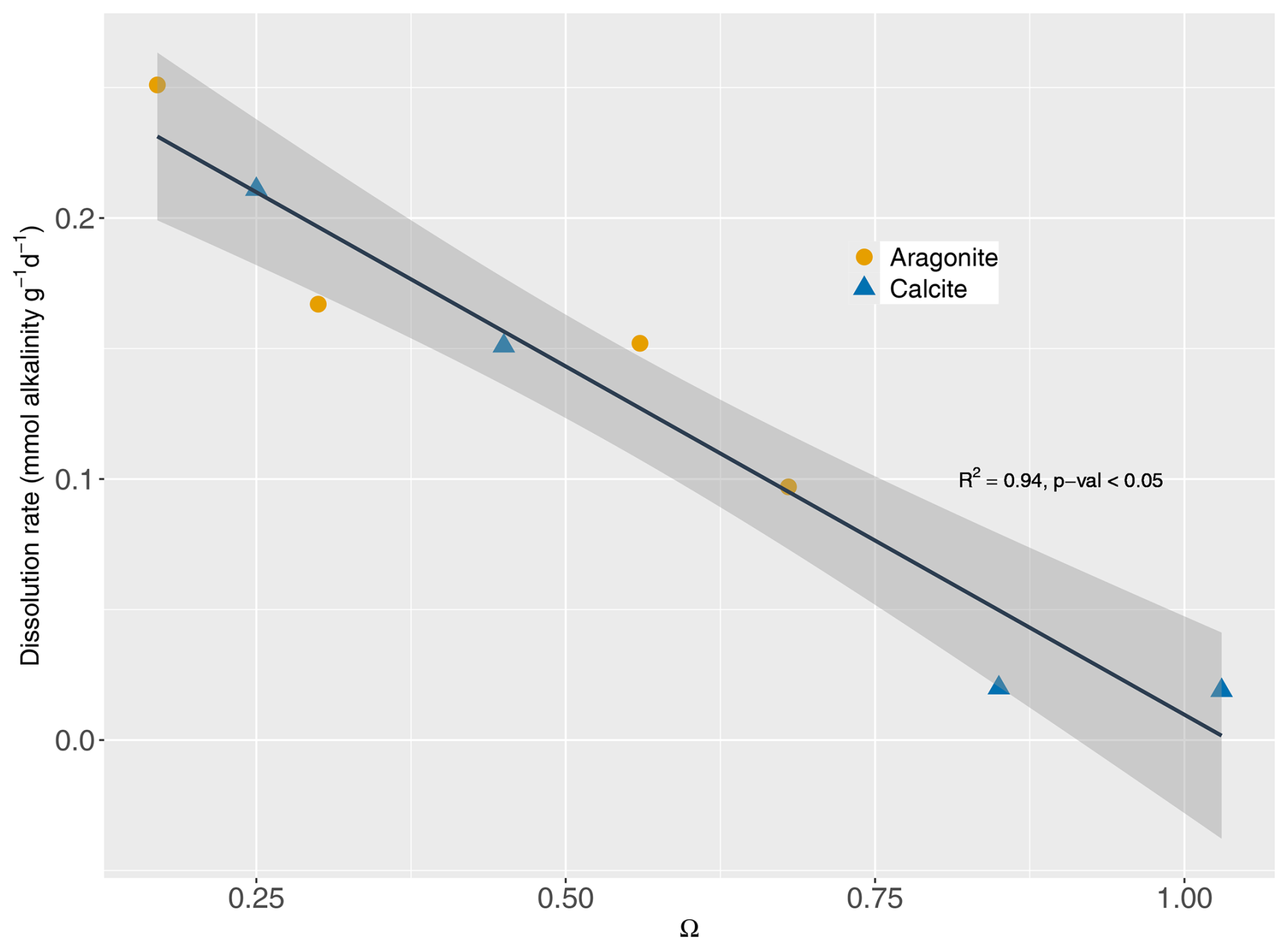
Figure 1Saturation state for aragonite and calcite vs. their respective dissolution rates (mmol alkalinity g d−1). Line shows the fit with n=1. R2: coefficient of determination. p value: statistical significance of the fit. Shaded area represents the 95 % confidence interval.
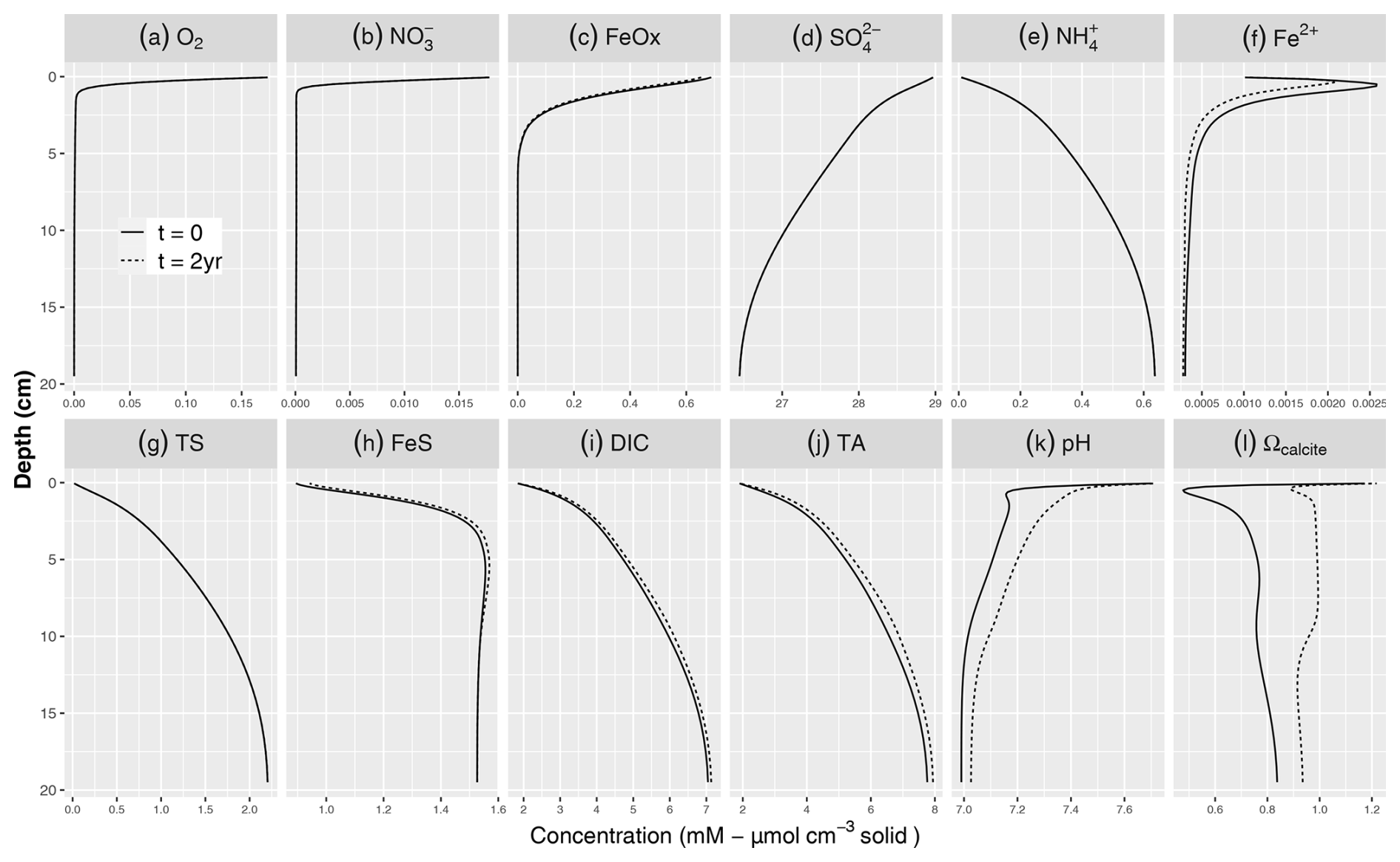
Figure 2Simulated baseline porewater profiles of (a) O2, (b) NO, (c) FeOx, (d) SO, (e) NH, (f) Fe2+, (g) TS, (h) FeS, (i) DIC, (j) TA, (k) pH, and (l) Ωcalcite with 8 % calcite addition. The solid lines denote the steady-state profiles prior to the addition of calcite to the top 2 cm, while the dashed lines indicate the simulated profiles 2 years after the mineral addition (some coincide with the solid lines and hence not visible). Dissolved constituents are in mmol L−1 porewater, while solids are given in µmol cm−3 solid phase.
3.2 Early diagenetic modeling
To assess the impact of sediment alkalinization through the addition of carbonate minerals to the sediment, early diagenetic model simulations were carried out that describe the spatiotemporal distribution of porewater and solid phase constituents.
Prior to the addition of minerals, the simulated porewater profiles show sequential depletion of electron acceptors with depth, with O2, NO, and reactive FeOx only present in the top 5 cm (solid lines in Fig. 2). SO is present throughout the depth range and only decreases by about 2.5 mM. As a result of mineralization reactions, the concentrations of reduced porewater constituents (TS (total sulfide), NH, Fe2+) increase with depth (Fig. 2). Reduced dissolved iron (Fe2+) peaked around 1 cm and then decreased with depth due to the precipitation of FeS, which in our baseline simulation limited the buildup of hydrogen sulfide (TS) and dissolved reduced iron (Fe2+). These porewater profiles reflect the sequential use of electron acceptors in the mineralization of organic matter, through aerobic respiration and anaerobic pathways such as denitrification, dissimilatory iron, and sulfate reduction. The dominant OM mineralization pathway was SO reduction, (78.9 %) followed by aerobic mineralization (17.2 %), denitrification, (3.9 %) and iron reduction (0.06 %).
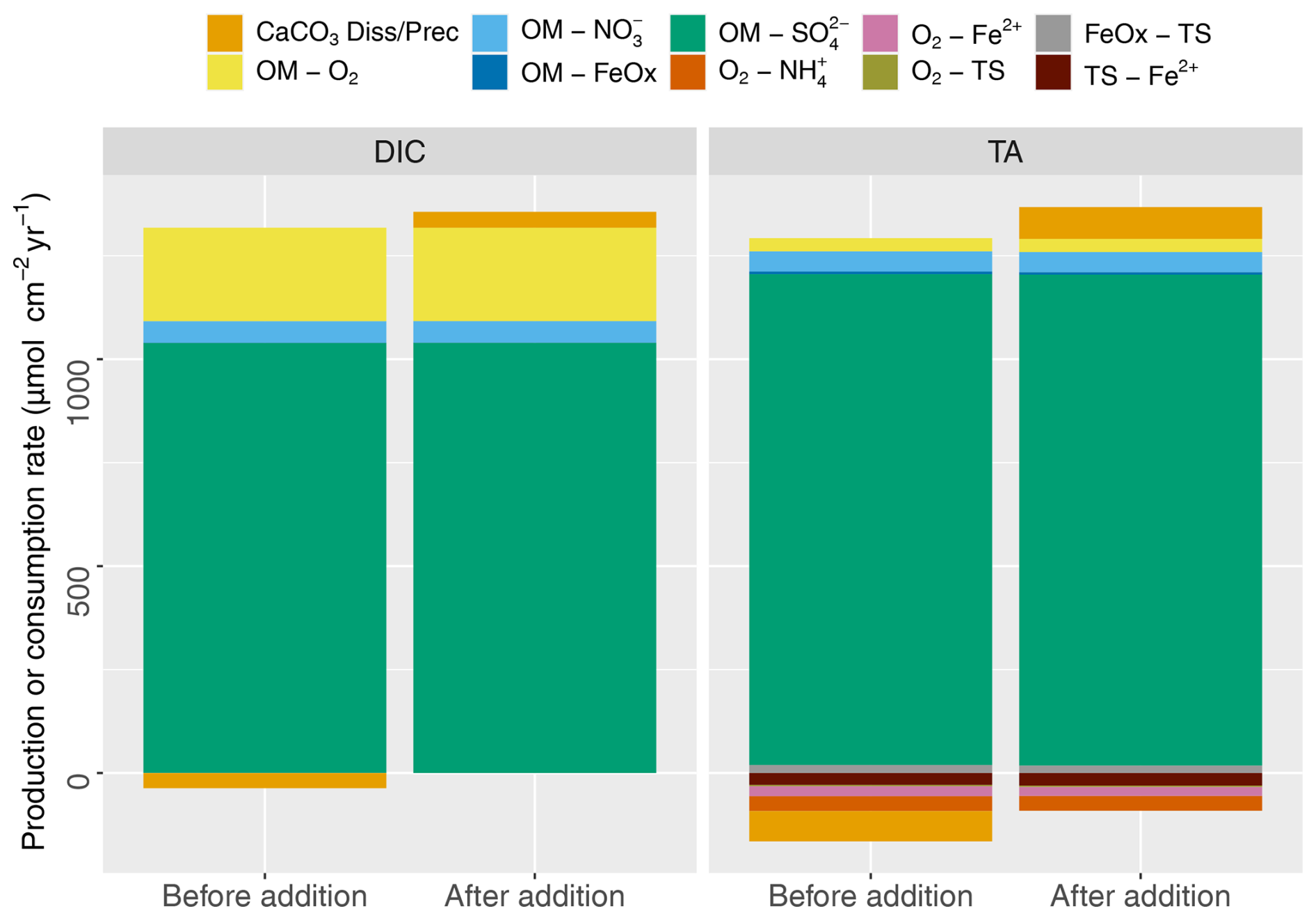
Figure 3DIC and TA production (positive) and consumption (negative) by different early diagenetic processes integrated over the entire model domain for steady state (t=0; before addition) and 2 years after the addition.
DIC and TA show pronounced concentration increases with depth, reflecting OM mineralization (Fig. 2i, j). The pH profile exhibits a rapid decrease in the top centimeter to about 7, showing the impact of CO2 production without the concurrent consumption of protons through aerobic mineralization (Table 3; Jahnke et al., 1994; Green et al., 2013). Below the oxic layer, active anaerobic OM mineralization pathways such as iron and sulfate reduction can generate considerable amounts of TA if the reaction products are not oxidized (Thomas et al., 2009). This was observed in the buildup of reduced species such as TS (discussed below), which limited the drop in pH and increased saturation state (from 0.5 to >0.8) in the deeper layers of the sediment (Fig. 2k, l). pH values were in the lower part of the range reported in Krumins et al. (2013), reflecting the lack of carbonates buffering the pH prior to the mineral additions. Sulfate reduction produced the majority of the TA and DIC (Fig. 3), consistent with the findings of Krumins et al. (2013), Brenner et al. (2016), and Gimenez (2018). Aerobic mineralization produced considerable amounts of DIC, in line with previous work (Thamdrup and Canfield, 2000), while generating less TA than anaerobic processes and mineral dissolution (Fig. 3), leading to a low porewater pH (Soetaert et al., 2007; Hofmann et al., 2011). DIC and TA production by denitrification and in particular dissimilatory iron reduction was substantially less. With a high dTA/dR value (Table 3), TS oxidation with FeOx also contributed to alkalinity production. Consumption of TA was dominated by secondary redox reactions that oxidize reduced species with O2. Due to fast kinetics, Fe2+ oxidation with O2 and nitrification and precipitation of FeS were the major TA-consuming reactions. Brenner et al. (2016) identified sulfide oxidation and nitrification as the dominant alkalinity-consuming processes in the shallow-water sediments in southern North Sea. This discrepancy may be due to the potentially substantial impact of sulfide oxidation on alkalinity dynamics in organically rich nearshore sediments (Krumins et al., 2013). The oxidation of reduced products of organic matter mineralization such as ammonium, hydrogen sulfide, and dissolved Fe2+ with O2 all lower alkalinity, albeit using different amounts of molecular oxygen. However, the oxidation of hydrogen sulfide with iron oxide as an electron donor produces a large amount of alkalinity that either accumulates if the reduced iron produced is captured in the sediment or is removed if the Fe2+ subsequently reacts with O2 in the oxic zone (see Table 3). Coupling of SO reduction with FeS precipitation has been shown to be an important mechanism for TA burial on continental shelves (Gustafsson et al., 2019). However, in our simulations, FeS formation was limited by Fe availability, which led to the buildup of hydrogen sulfide in the porewater.

Figure 4Temporal evolution of depth-averaged TA after addition of bioaragonite and biocalcite (a) and saturation states (b) in the baseline simulation. Concentrations and saturation states are averaged over the top 10 cm of sediment. Colors show the type of mineral added. Solid and dashed lines show the addition of 16 % and 8 %, respectively.
3.2.1 Effect of carbonate mineral additions
After adding carbonate minerals, the concentration profiles of primary redox species remained largely unchanged (Fig. 2). This is because the organic matter mineralization rate expressions (Table 3) do not contain dependencies on the carbonate system or pH, as is common in early diagenetic models (e.g., Jourabchi et al., 2008; Morse and Mackenzie, 1990). Mineral addition slightly decreased Fe2+ while increasing FeS concentrations. This is due to TA production due to carbonate dissolution, which elevates the pH and increases the saturation state of FeS (Table 3), promoting FeS formation, which eventually can lead to the long-term removal of reduced sulfur through pyrite burial (Hu and Cai, 2011a)
The added carbonate minerals started to dissolve in our model, producing one DIC and two TA per carbon (Burdige et al., 2010; Zeebe and Wolf-Gladrow, 2001) so that both the pH (Fig. 2k) and porewater saturation state (Figs. 2l and 4b) initially increased (Cai et al., 2011). After 2 years, CO2 production still lowered pH in the oxic layer and prevented full saturation. However, Ω was always above 0.9 and reached 1 at and below a depth of approximately 2.5 cm, and subsequent decrease was observed below 12 cm. Following the addition of carbonates, the extent of the undersaturated conditions was constrained to the top 2 cm only, while in the natural buffering scenario reported in Krumins et al. (2013) porewater remained undersaturated in the top 10 cm. This demonstrates the potential of OAE in mitigating surface sediment acidification and providing potential benefits for calcifiers living in the upper sediment layers. As mineral addition counteracts porewater acidification, it generates biogeochemical conditions conducive to the survival of juvenile bivalves.
Our results are in line with the literature showing that the main alkalinity-producing processes were calcite mineral dissolution, sulfate reduction, denitrification, and sulfide oxidation with iron oxides (Krumins et al., 2013; Berelson et al., 2007). Although organic matter mineralization reactions generate alkalinity, they have lower dTA / dDIC ratios than mineral dissolution on a per carbon basis (Table 3), and the TA-producing mineralization reactions are mostly coupled to TA-consuming reoxidation reactions. Therefore, the net effect of (some) mineralization pathways on TA production can be limited (Krumins et al., 2013). For example, the amount of TA generated by benthic denitrification was almost balanced out by NH oxidation (Fig. 3), which results in the small effect of N cycling on alkalinity. This is in line with previous studies showing strong coupling between oxidation/reduction of N in the marine environment (Middelburg et al., 1996) and its weak impact on buffering capacity of the overlying water (Hu and Cai, 2011b). Since reoxidation reactions are dependent on the availability of O2, enhanced biological mixing can also increase TA consumption, as previously demonstrated by Rao et al. (2014) for Fe2+ oxidation by O2. Although total production of TA was around 1300 µmol cm−2 yr−1 (Fig. 3, baseline with biocalcite), total TA fluxes from biocalcite were slightly lower, which points to the importance of consumption of TA in the oxic layer as previously reported in Krumins et al. (2013). Finally, before mineral addition, we observed a small production of calcite at the top of the sediment due to oversaturated conditions in overlying water. After the addition, however, the added mineral dissolved in deeper layers, which led to net dissolution and generation of alkalinity in the sediment column.
3.2.2 Temporal evolution of buffering with different minerals and amounts
Longer-term simulations (50 years) were performed to investigate the evolution of the buffering with time, considering different amounts and types of minerals added. We focused on the biogeochemical conditions in the top 10 cm of sediment, because the surficial sediment is most relevant for juvenile bivalves impacted by acidification. Figure 4 shows the temporal evolution of TA and saturation state following the addition of minerals to the steady-state conditions at t=0. Depth-averaged TA concentrations in the top 10 cm peaked around the second year after addition, with a significant drop starting around year 10 for the 8% mineral addition and around year 15 for the 16 % addition and then a return to steady-state levels after more than 30 years (Fig. 4a). Doubling the amount of mineral added generated slightly higher TA and extended the length of the peak buffering period. The simulation of aragonite addition produced higher alkalinity concentrations than that of calcite by approximately 0.3 mM at the peak time, reflecting its higher solubility. However, TA levels in aragonite application dropped faster after approximately 10 and 15 years and approached the initial premineral application levels after about 30 and 40 years, for 8 % and 16 % additions, respectively. In contrast, following the addition of calcite, TA concentrations remained elevated over longer time periods. Averaging over 4 cm (not shown) yielded the same patterns (with slightly lower TA concentrations) as shown in Fig. 4.
Being less stable than calcite, aragonite had a slightly faster dissolution rate constant and a larger solubility product (Burdige et al., 2010). Therefore, it generated higher TA concentrations and benthic fluxes early on. Because of its higher solubility, it initially took aragonite slightly longer to reach saturation than calcite (i.e., orange curve reaches maximum slightly after the blue one). As the rate of dissolution is set to be proportional not only to the degree of undersaturation but also the carbonate mineral concentration (Eq. 2), doubling the mineral addition had a more pronounced effect on aragonite, which approached saturation more slowly than calcite (Fig. 4b). Burial of carbonate minerals below 10 cm led to a faster decrease in TA in aragonite due to its higher dissolution. Initial conditions in 50-year simulations with calcite had a higher saturation state than aragonite implementation. Therefore, after the impact of addition was buried, calcite leveled at a higher saturation state and TA than aragonite.
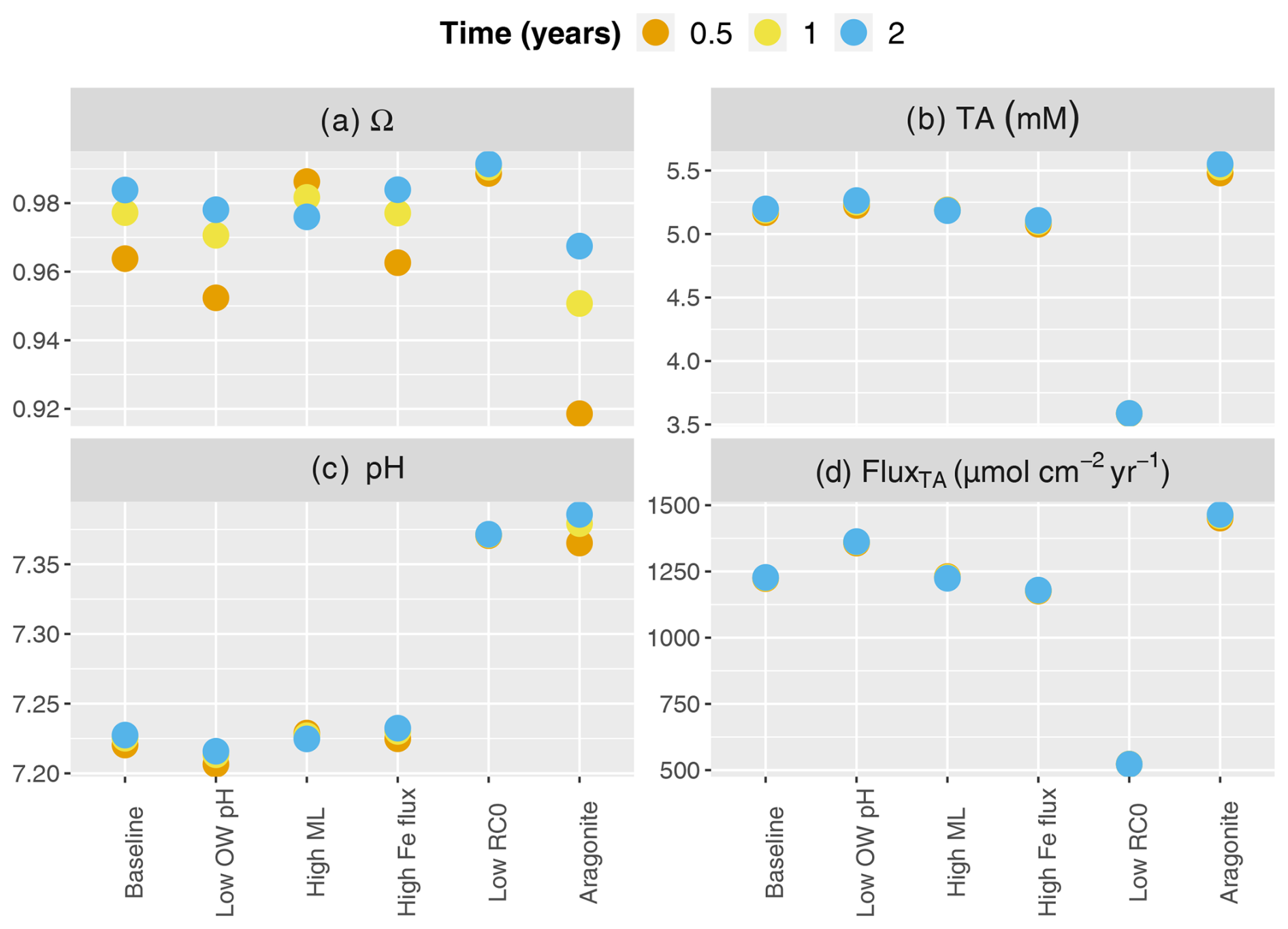
Figure 5Buffering response after carbonate mineral additions for a range of environmental conditions. Concentrations are averaged over the top 10 cm. Scenarios listed on the x axis (from left to right) represent the baseline simulation the baseline with lower overlying water pH representing the ocean acidification scenario (7.8), the baseline with the deeper mixed layer (10 cm), the baseline with a higher Fe flux (10 µmol cm−2 yr−1), the baseline with lower (200 µmol cm−3 yr−1), and the baseline with aragonite instead of calcite.
3.2.3 Sensitivity analyses
The impact of mineral additions on the generation of alkalinity depends not only on the type of mineral added, but also on which early diagenetic processes dominate (Fig. 3). The relative magnitude of these processes depends on a number of factors, including the rate of organic matter mineralization, the extent of bioturbation, the deposition fluxes of potential electron acceptors such as iron oxyhydroxides, and the composition of the overlying water. Varying these factors in our model, we explore their impact on both the environmental conditions in the surficial sediment and the impact on benthic alkalinity fluxes.
In our simulations, under all scenarios, mineral saturation states reached similar values at the end of the 2 years with a low scenario generating the highest values (blue dots in Fig. 5a). However, saturation was reached fastest in the deeper mixed-layer scenario, which then decreased by years 1 and 2. Baseline, high FeOx flux and low scenarios showed a similar time course, while the addition of aragonite instead of calcite exhibited the slowest increase in saturation state (Fig. 5a). Depth-averaged TA concentrations were highest following the addition of aragonite (Fig. 5b). Deepening the bioturbation layer did not have much of an impact. The high FeOx flux scenario produced slightly lower TA than the baseline, and the lowest amounts of alkalinity were produced by the low scenario. pH increased the most and fastest in the aragonite implementation, followed by low conditions (Fig. 5c). Although the deep mixed-layer scenario increased pH faster than the baseline and high-FeOx-flux scenarios, these three scenarios converged to a similar pH to the impact of mineral dissolution developed. The highest benthic TA fluxes were generated during the dissolution of aragonite, while the lowest values were produced in the low scenario (Fig. 5d). Baseline and deeper bioturbation layer scenarios produced very similar results, while the high-FeOx-flux scenario produced slightly lower TA fluxes. Lowering overlying water pH led to decreases in saturation state and pH while increasing concentration and flux of alkalinity.
A lower pH in the overlying water that represents ocean acidification led to a lower pH in the porewater and less saturated conditions in surface sediments (Fig. 5). This promoted mineral dissolution and therefore led to increased TA production and flux to the overlying water. This demonstrates the sensitivity of buffering to the overlying water conditions. Deeper bioturbation led to faster downward mixing and subsequent dissolution of the added mineral, which then led to a rapid increase in TA, pH, and saturation state (also seen in dpH/dTA; Fig. 4). Higher mixing led to faster transport of minerals inducing enhanced dissolution in year 0.5 (i.e., Fig. 5; higher buffering after 0.5 than 2 years after mineral addition). However, faster transport extended the depth of buffering and diluted dissolution products, which caused a slight decrease in pH and saturation state after 2 years at the top 10 cm (Fig. 5). Increased FeOx input to the sediment led to enhanced FeOx reduction, which produced TA. However, increased reoxidation of Fe2+ led to decreased availability of O2 and consumption of TA. This balanced out the additional TA produced through FeOx reduction and led to the additional net consumption of ∼46 µmol cm−2 yr−1 of TA. Overall, increased iron oxide supply led to slight decreases in TA concentrations and fluxes and small increases in pH, showing an effect similar to decreasing the OM mineralization rate (Fig. 5). TA production and flux were significantly lower in the scenario with a low OM reaction rate (Fig. 5d), which showed the critical impact of OM mineralization on TA dynamics. The lower rate of OM mineralization decreases TA production, both through the reduction of overall mineralization rates and the reduction of the relative contribution of anaerobic mineralization pathways like dissimilatory sulfate reduction. The low rate of aerobic mineralization also led to reduced H+ production, which in turn slows carbonate dissolution (Morse and Mackenzie, 1990). However, even with lower dissolution, this situation led to higher pH than the baseline, suggesting that the artificial buffering might not be needed to maintain saturated conditions in environments where OM reactivity is low. In deeper layers where the surficial mineral addition is not as effective, TA and mineral saturation states will be lower due to lower anaerobic TA production. Notably, the effect of changing the OM mineralization rate is not linear, and halving the rate reduced the benthic alkalinity flux by a factor of about 2.5. In our simulations, the lower OM mineralization rate led to depth-integrated reduction by 29 % for aerobic mineralization, 19 % for denitrification, 55 % for sulfate reduction, and 380 % for mineral dissolution. Aside from the impact on the partitioning of OM mineralization between metabolic pathways, this mainly reflects that changes in mineralization rates lead to differences in the porewater conditions, which in turn alter the rate of mineral dissolution/precipitation. Due to the higher solubility and hence larger degree of undersaturation, initial aragonite dissolution was faster than calcite. Hence, aragonite produced the largest TA in 2 years, generated the highest benthic TA fluxes, and increased the pH the most. The addition of aragonite produced more buffering than the scenario with lower OM mineralization, which shows the potential of artificial buffering overcoming the acidification impact due to OM mineralization.
3.3 Limitations and challenges
Our model demonstrates the potential for coastal sediment alkalinization to improve the biogeochemical conditions for juvenile calcifying organisms living in surficial sediments. However, alkalinity fluxes are sensitive to overlying water conditions; temporal changes in overlying water should be considered. These changes include the impact of short-term diel patterns and small-scale spatial heterogeneity on benthic fluxes (Gadeken et al., 2023), seasonal changes such as temperature (Rao et al., 2014), and long-term changes such as anthropogenic impacts (Pacella et al., 2018; e.g., eutrophication, Cai et al., 2011). In addition, in particular in nearshore shallow-water environments, one may have to take into consideration the input of groundwater (Cyronak et al., 2013a), advective flow through sand ripples (Huettel et al., 1998), and other factors driving advective flow in marine sediments (Santos et al., 2012), which can impact sediment biogeochemistry and alkalinity fluxes. Furthermore, the prediction of the time course of buffering by mineral dissolution may need to consider dissolution rate expressions that account for changes in mineral surface areas, rather than bulk concentrations (Morse et al., 2007), and the potential for washing away or enhancing dissolution (Meysman and Montserrat, 2017) of minerals deposited to the sediment in high-energy environments.
Our results indicate a lasting buffering effect of mineral additions. This finding depends on the kinetics of the mineral dissolution rate, in particular the reaction order. Given the range of observations (Adkins et al., 2021; Subhas et al., 2018), the temporal evolution of the dissolution rates in the field needs to be explored further. In addition, our model assumes that the minerals are mixed into the upper 2 cm of sediment, matching recent field manipulation experiments in Yaquina Bay. However, an important consideration is how the minerals are applied to the surface sediment. For example, a simple “top-dressing” approach would involve less disturbance to the sediment and less labor than surface raking, but it may be washed away more easily and react less readily with the metabolic acids produced in the underlying sediment. Thus, optimal mineral application and subsequent effects clearly require field trials across a range of hydrodynamic settings.
3.4 Potential impact of benthic fluxes on marine CO2 uptake
Benthic alkalinity fluxes are critical for water column C dynamics, especially in shallow-water environments where water column and atmospheric interactions are more sensitive to processes occurring in the sediment (Brenner et al., 2016). The significant decrease in shells due to overharvesting has likely altered alkalinity cycling in estuarine waters, such as the Chesapeake Bay (Waldbusser et al., 2013). One outcome of this benthic–pelagic connection is the impact of benthic fluxes on oceanic CO2 uptake, which was estimated to have a significant influence in the North Sea (Thomas et al., 2009), and the potential for shallow-water sediment alkalinization to alter overlying water.
We estimated the potential impact of enhanced sediment TA fluxes on atmospheric CO2 uptake in Yaquina Bay, OR (USA), using the diagenetic model output (baseline scenario) in a simple box model during the peak buffering period (∼2 years after addition). The bay was found to be net heterotrophic, with tidal exchange and net respiration in the water column impacting air–sea CO2 flux the most. The impact of sediment buffering on the air–sea CO2 was estimated as the difference in atmospheric CO2 fluxes between before and after the mineral addition. Mineral addition resulted in a TA flux enhancement of ∼126 µmol cm−2 yr−1 and a decrease in CO2 flux by approximately 58 µmol cm−2 yr−1, which was comparable to previous studies (Brenner et al., 2016). Scaling to a 10 m water depth (or per liter of water), this stoichiometrically estimated marine carbon dioxide removal (mCDR) due to the application and subsequent dissolution of carbonate minerals by the sediments results in a similar amount of mCDR to the current estimates of the anthropogenic C (Canth) concentration in the US Pacific coast waters, 30–60 µmol L−1 (Feely et al., 2016; Pacella et al., 2024). It is important to note three key points in this estimate: first, the uptake of Canth by marine waters from anthropogenic activities results in an increase in DIC without any concurrent change in alkalinity and thus acidification. Second, alkalinity enhancement shifts the carbonate speciation and allows increased uptake of atmospheric CO2 by marine waters. Thus, ocean alkalinity enhancement increases the total amount of Canth in seawater. Third, however, Canth and DIC both increase with OAE; the mineral dissolution due to sediment respiration results in an increase in the TA : DIC ratio, thus providing the mCDR benefit of atmospheric removal as well as mitigating existing acidification impacts. The annual mCDR effect modeled here, while notable compared to the total anthropogenic impact, is minuscule compared to the total air–sea exchange flux, which is driven by tidal exchange of DIC and alkalinity between the bay and the coastal ocean and biogeochemical processes in the water column. The signal-to-noise ratio of this potentially significant effect also highlights the existing measurement challenges in accurately discerning OAE effects within coastal systems.
Our study successfully combines lab experiments and modeling to investigate the impact of coastal alkalinity enhancement, points to critical aspects to be considered, and presents the potential of carbonate additions as a mitigation strategy for the effects of climate change in coastal settings. We demonstrated that addition of minerals increased pH and saturation state of the sediment over the time span of a few months and had the potential to persist over 30–40 years, after which subsequent additions might be needed to sustain buffering. During the peak buffering period, porewater in the top 10 cm of the sediment was almost fully saturated with respect to the mineral added, which implies favorable conditions for benthic calcifying organisms. We showed that two main pathways of producing TA in the sediment were sulfate reduction and mineral dissolution, with sulfate reduction having a larger impact. We also demonstrated that the type of mineral was an important factor affecting benthic fluxes and that the application of an increased amount of minerals was effective in extending the duration rather than the strength of the buffering.
Our analysis suggests that buffering through mineral additions to coastal sediments can establish biogeochemical conditions that are conducive to the growth and development of calcifying benthic organisms. However, the effect of mineral additions can vary substantially between environments. Sensitivity analyses demonstrated that the OM mineralization had a significant impact on mineral dissolution, pH, and mineral saturation. The type of mineral added also showed a significant effect, with aragonite producing more TA and higher pH. Increased mixing depth led to a faster increase in saturation state, while increased iron flux caused reduction in TA concentration and benthic fluxes. Overall, the addition of minerals with higher dissolution rates would be most effective especially in environments with high OM degradation and undersaturated water conditions. Similarly, the type of mineral and the environmental conditions affect mCDR potential through benthic fluxes. Importantly, the implementation of OAE can lead to unforeseen effects, including ecological feedback such as changes in species composition (Bach et al., 2019; Köhler et al., 2013), and the effectiveness of carbonate additions may depend strongly on site characteristics. Thus, studies should combine field, lab, and modeling components (Meysman and Montserrat, 2017) to improve predictive capabilities and decrease uncertainty.
The reaction transport model can be found at https://doi.org/10.5281/zenodo.14751492 (Biçe and Meile, 2024).
Data for this article are available in Myers (2022) (https://ir.library.oregonstate.edu/concern/graduate_thesis_or_dissertations/br86bb964).
The supplement related to this article is available online at: https://doi.org/10.5194/bg-22-641-2025-supplement.
Lab experiments were designed by GW and TMS and carried out by TMS with the guidance of GW. KB developed the numerical model with the guidance of CM and carried out the simulations. KB prepared the initial draft of the manuscript with significant input from CM, TMS, and GW.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors would like to thank the ClimateWorks Foundation for funding and Jurjen Rooze for input on the model implementation.
This research has been supported by the ClimateWorks Foundation (grant no. 20-1534 to George G. Waldbusser and Christof Meile), and unpublished data used to estimate model end members was supported by Oregon Ocean Science Trust (grant no. 22-287 to George G. Waldbusser).
This paper was edited by Tyler Cyronak and reviewed by three anonymous referees.
Adkins, J. F., Naviaux, J. D., Subhas, A. V., Dong, S., and Berelson, W. M.: The dissolution rate of CaCO3 in the ocean, Annu. Rev. Mar. Sci., 13, 57–80, 2021.
Aller, R. C.: The effects of macrobenthos on chemical properties of marine sediment and overlying water, in: Animal-sediment relations: the biogenic alteration of sediments, Boston, MA, Springer US, 53–102 , 1982.
Bach, L. T., Gill, S. J., Rickaby, R. E., Gore, S., and Renforth, P.: CO2 removal with enhanced weathering and ocean alkalinity enhancement: potential risks and co-benefits for marine pelagic ecosystems, Front. Clim., 1, 7, https://doi.org/10.3389/fclim.2019.00007, 2019.
Bandstra, L., Hales, B., and Takahashi, T.: High-frequency measurements of total CO2: Method development and first oceanographic observations, Mar. Chem., 100, 24–38, 2006.
Barton, A., Hales, B., Waldbusser, G. G., Langdon, C., and Feely, R. A.: The Pacific oyster, Crassostrea gigas, shows negative correlation to naturally elevated carbon dioxide levels: Implications for near-term ocean acidification effects, Limnol. Oceanogr., 57, 698–710, 2012.
Berelson, W. M., Balch, W. M., Najjar, R., Feely, R. A., Sabine, C., and Lee, K.: Relating estimates of CaCO3 production, export, and dissolution in the water column to measurements of CaCO3 rain into sediment traps and dissolution on the sea floor: A revised global carbonate budget, Global Biogeochem. Cy., 21, GB1024, https://doi.org/10.1029/2006GB002803, 2007.
Berner, R. A.: Early diagenesis: a theoretical approach, 241 pp., Princeton University Press, ISBN 9780691082608, 1980.
Biçe, K. and Meile, C.: 1-D reactive transport model for coastal sediment alkalinization, Zenodo [code], https://doi.org/10.5281/zenodo.14751492, 2024.
Boudreau, B. P.: On the equivalence of nonlocal and radial-diffusion models for porewater irrigation, J. Marine Res., 42, 731–735, 1984.
Boudreau, B. P.: The diffusive tortuosity of fine-grained unlithified sediments, Geochim. Cosmochim. Ac., 60, 3139–3142, 1996.
Boudreau, B. P.: Diagenetic models and their implementation, 414 pp., Vol. 505, Berlin, Springer, ISBN 9783642643996, 1997.
Brenner, H., Braeckman, U., Le Guitton, M., and Meysman, F. J. R.: The impact of sedimentary alkalinity release on the water column CO2 system in the North Sea, Biogeosciences, 13, 841–863, https://doi.org/10.5194/bg-13-841-2016, 2016.
Brown, C. A. and Ozretich, R. J.: Coupling between the coastal ocean and Yaquina Bay, Oregon: Importance of oceanic inputs relative to other nitrogen sources, Estuar. Coast., 32, 219–237, 2009.
Burdige, D. J.: Preservation of organic matter in marine sediments: controls, mechanisms, and an imbalance in sediment organic carbon budgets?, Chem. Rev., 107, 467–485, 2007.
Burdige, D. J., Hu, X., and Zimmerman, R. C.: The widespread occurrence of coupled carbonate dissolution/reprecipitation in surface sediments on the Bahamas Bank, Am. J. Sci., 310, 492–521, 2010.
Cai, W. J., Hu, X., Huang, W. J., Murrell, M. C., Lehrter, J. C., Lohrenz, S. E., Chou, W. C., Zhai, W., Hollibaugh, J. T., Wang, Y., Zhao, P., Guo, X., Gundersen, K., Dai, M., and Gong, G. C.: Acidification of subsurface coastal waters enhanced by eutrophication, Nat. Geosci., 4, 766–770, https://doi.org/10.1038/ngeo1297, 2011.
Cai, W. J., Huang, W. J., Luther III, G. W., Pierrot, D., Li, M., Testa, J., Xue, M., Joesoef, A., Mann, R., Brodeur, J., Xu, Y. Y., Chen, B., Hussain, N., Waldbusser, G. G., Cornwell, J., and Kemp, W. M.: Redox reactions and weak buffering capacity lead to acidification in the Chesapeake Bay, Nat. Commun., 8, 1–12, https://doi.org/10.1038/s41467-017-00417-7, 2017.
Cyronak, T., Santos, I. R., Erler, D. V., and Eyre, B. D.: Groundwater and porewater as major sources of alkalinity to a fringing coral reef lagoon (Muri Lagoon, Cook Islands), Biogeosciences, 10, 2467–2480, https://doi.org/10.5194/bg-10-2467-2013, 2013a.
Cyronak, T., Santos, I. R., McMahon, A., and Eyre, B. D.: Carbon cycling hysteresis in permeable carbonate sands over a diel cycle: Implications for ocean acidification, Limnol. Oceanogr., 58, 131–143, 2013b.
D'Andrea, A. F. and DeWitt, T. H.: Geochemical ecosystem engineering by the mud shrimp Upogebia pugettensis (Crustacea: Thalassinidae) in Yaquina Bay, Oregon: Density-dependent effects on organic matter remineralization and nutrient cycling, Limnol. Oceanogr., 54, 1911–1932, 2009.
Dickson, A. G.: An exact definition of total alkalinity and a procedure for the estimation of alkalinity and total inorganic carbon from titration data, Deep-Sea Res. Pt. I, 28, 609–623, 1981.
Dickson, A. G.: Standard potential of the reaction: AgCl (s)+ 12H2 (g)= Ag (s)+ HCl (aq), and and the standard acidity constant of the ion HSO4− in synthetic sea water from 273.15 to 318.15 K, J. Chem. Thermodyn., 22, 113–127, 1990.
Edmond, J. M. and Gieskes, J. M. T. M.: On the calculation of the degree of saturation of sea water with respect to calcium carbonate under in situ conditions, Geochim. cosmochim. Ac., 34, 1261–1291, 1970.
Egleston, E. S., Sabine, C. L., and Morel, F. M.: Revelle revisited: Buffer factors that quantify the response of ocean chemistry to changes in DIC and alkalinity, Global Biogeochem. Cy., 24, GB1002, https://doi.org/10.1029/2008GB003407, 2010.
Feely, R. A., Alin, S. R., Carter, B., Bednaršek, N., Hales, B., Chan, F., Hill, T. M., Gaylord, B., Sanford, E., Byrne, R. H., Sabine, C. L., Greeley, D., and Juranek, L.: Chemical and biological impacts of ocean acidification along the west coast of North America, Estuar. Coast. Shelf Sci., 183, 260–270, 2016.
Fennel, K., Long, M. C., Algar, C., Carter, B., Keller, D., Laurent, A., Mattern, J. P., Musgrave, R., Oschlies, A., Ostiguy, J., Palter, J. B., and Whitt, D. B.: Modelling considerations for research on ocean alkalinity enhancement (OAE), in: Guide to Best Practices in Ocean Alkalinity Enhancement Research, edited by: Oschlies, A., Stevenson, A., Bach, L. T., Fennel, K., Rickaby, R. E. M., Satterfield, T., Webb, R., and Gattuso, J.-P., Copernicus Publications, State Planet, 2-oae2023, 9, https://doi.org/10.5194/sp-2-oae2023-9-2023, 2023.
Ferderer, A., Chase, Z., Kennedy, F., Schulz, K. G., and Bach, L. T.: Assessing the influence of ocean alkalinity enhancement on a coastal phytoplankton community, Biogeosciences, 19, 5375–5399, https://doi.org/10.5194/bg-19-5375-2022, 2022.
Fuhr, M., Wallmann, K., Dale, A. W., Kalapurakkal, H. T., Schmidt, M., Sommer, S., Deusner, C., Spiegel, T., Kowalski, J., and Geilert, S.: Alkaline mineral addition to anoxic to hypoxic Baltic Sea sediments as a potentially efficient CO2-removal technique, Front. Clim., 6, 1338556, https://doi.org/10.3389/fclim.2024.1338556, 2024.
Gadeken, K. J., Lockridge, G., and Dorgan, K. M.: An in situ benthic chamber system for improved temporal and spatial resolution measurement of sediment oxygen demand, Limnol. Oceanogr.-Methods, 21, 645–655, 2023.
Gimenez, I., Waldbusser, G. G., and Hales, B.: Ocean acidification stress index for shellfish (OASIS): Linking Pacific oyster larval survival and exposure to variable carbonate chemistry regimes, Elem. Sci. Anth., 6, 51, https://doi.org/10.1525/elementa.306, 2018.
Goldenberg, S. U., Riebesell, U., Brüggemann, D., Börner, G., Sswat, M., Folkvord, A., Couret, M., Spjelkavik, S., Sánchez, N., Jaspers, C., and Moyano, M.: Early life stages of fish under ocean alkalinity enhancement in coastal plankton communities, Biogeosciences, 21, 4521–4532, https://doi.org/10.5194/bg-21-4521-2024, 2024.
Green, M. A., Waldbusser, G. G., Reilly, S. L., Emerson, K., and O'Donnell, S.: Death by dissolution: sediment saturation state as a mortality factor for juvenile bivalves, Limnol. Oceanogr., 54, 1037–1047, 2009.
Green, M. A., Waldbusser, G. G., Hubazc, L., Cathcart, E., and Hall, J.: Carbonate mineral saturation state as the recruitment cue for settling bivalves in marine muds, Estuar. Coast., 36, 18–27, 2013.
Gustafsson, E., Hagens, M., Sun, X., Reed, D. C., Humborg, C., Slomp, C. P., and Gustafsson, B. G.: Sedimentary alkalinity generation and long-term alkalinity development in the Baltic Sea, Biogeosciences, 16, 437–456, https://doi.org/10.5194/bg-16-437-2019, 2019.
Hales, B., Takahashi, T., and Bandstra, L.: Atmospheric CO2 uptake by a coastal upwelling system, Global Biogeochem. Cy., 19, Gb1009, https://doi.org/10.1029/2004gb002295, 2005.
Hales, B., Suhrbier, A., Waldbusser, G. G., Feely, R. A., and Newton, J. A.: The carbonate chemistry of the “fattening line,” Willapa Bay, 2011–2014, Estuar. Coast., 40, 173–186, 2017.
Hangx, S. J. and Spiers, C. J.: Coastal spreading of olivine to control atmospheric CO2 concentrations: A critical analysis of viability. Int. J. Greenh. Gas Con., 3, 757–767, 2009.
Hartmann, J., West, A. J., Renforth, P., Köhler, P., Rocha, C. L. D. L., Wolf-Gladrow, D. A., Dürr, H. H., and Scheffran, J.: Enhanced chemical weathering as a geoengineering strategy to reduce atmospheric carbon dioxide, supply nutrients, and mitigate ocean acidification, Rev. Geophys., 51, 113–149, 2013.
Hofmann, A. F., Soetaert, K., Middelburg, J. J., and Meysman, F. J.: AquaEnv: An Aquatic Acid–Base Modelling Environment in R, Aquat. Geochem., 16, 507–546, 2010.
Hofmann, G. E., Smith, J. E., Johnson, K. S., Send, U., Levin, L. A., Micheli, F., Paytan, A., Price, N. N., Peterson B., Takeshita, Y., Matson, P. G., Crook, E. D., Kroeker, K. J., Gambi, M. C., Rivest, E. B., Frieder, C. A., Yu, P. C., and Martz, T. R.: High-frequency dynamics of ocean pH: A multi-ecosystem comparison, PLoSONE, 6, e28983, https://doi.org/10.1371/journal.pone.0028983, 2011.
Hu, X. and Cai, W. J.: An assessment of ocean margin anaerobic processes on oceanic alkalinity budget, Global Biogeochem. Cy., 25, GB3003, https://doi.org/10.1029/2010GB003859, 2011a.
Hu, X. and Cai, W. J.: The impact of denitrification on the atmospheric CO2 uptake potential of seawater, Mar. Chem., 127, 192–198, 2011b.
Huettel, M., Ziebis, W., Forster, S., and Luther Iii, G. W.: Advective transport affecting metal and nutrient distributions and interfacial fluxes in permeable sediments, Geochim. Cosmochim. Ac., 62, 613–631, 1998.
Jahnke, R. A., Craven, D. B., and Gaillard, J. F.: The influence of organic matter diagenesis on CaCO3 dissolution at the deep-sea floor, Geochim. Cosmochim. Ac., 58, 2799–2809, 1994.
Jourabchi, P., Van Cappellen, P., and Regnier, P.: Quantitative interpretation of pH distributions in aquatic sediments: A reaction-transport modeling approach, Am. J. Sci., 305, 919–956, 2005.
Jourabchi, P., Meile, C., Pasion, L. R., and Van Cappellen, P.: Quantitative interpretation of pore water O2 and pH distributions in deep-sea sediments, Geochim. Cosmochim. Ac., 72, 1350–1364, 2008.
Köhler, P., Abrams, J. F., Völker, C., Hauck, J., and Wolf-Gladrow, D. A.: Geoengineering impact of open ocean dissolution of olivine on atmospheric CO2, surface ocean pH and marine biology, Environ. Res.Lett., 8, 014009, https://doi.org/10.1088/1748-9326/8/1/014009, 2013.
Krumins, V., Gehlen, M., Arndt, S., Van Cappellen, P., and Regnier, P.: Dissolved inorganic carbon and alkalinity fluxes from coastal marine sediments: model estimates for different shelf environments and sensitivity to global change, Biogeosciences, 10, 371–398, https://doi.org/10.5194/bg-10-371-2013, 2013.
Lueker, T. J., Dickson, A. G., and Keeling, C. D.: Ocean pCO2 calculated from dissolved inorganic carbon, alkalinity, and equations for K1 and K2: validation based on laboratory measurements of CO2 in gas and seawater at equilibrium, Mar. Chem., 70, 105–119, 2000.
Luff, R. and Wallmann, K.: Fluid flow, methane fluxes, carbonate precipitation and biogeochemical turnover in gas hydrate-bearing sediments at Hydrate Ridge, Cascadia Margin: numerical modeling and mass balances, Geochim. Cosmochim. Ac., 67, 3403–3421, 2003.
Meile, C. and Van Cappellen, P.: Global estimates of enhanced solute transport in marine sediments, Limnol. Oceanogr., 48, 777–786, 2003.
Meysman, F. J. and Montserrat, F.: Negative CO2 emissions via enhanced silicate weathering in coastal environments, Biol. Lett., 13, 20160905, https://doi.org/10.1098/rsbl.2016.0905, 2017.
Middelburg, J. J., Soetaert, K., Herman, P. M., and Heip, C. H.: Denitrification in marine sediments: A model study, Global Biogeochem. Cy., 10, 661–673, 1996.
Middelburg, J. J., Soetaert, K., and Herman, P. M.: Empirical relationships for use in global diagenetic models, Deep-Sea Res. Pt. I, 44, 327–344, 1997.
Millero, F. J.: Carbonate constants for estuarine waters, Mar. Freshwater Res., 61, 139–142, 2010.
Montserrat, F., Renforth, P., Hartmann, J., Leermakers, M., Knops, P., and Meysman, F. J.: Olivine dissolution in seawater: implications for CO2 sequestration through enhanced weathering in coastal environments, Environ. Sci. Technol., 51, 3960–3972, 2017.
Morris, A. W. and Riley, J. P.: The bromide/chlorinity and sulphate/chlorinity ratio in sea water, Deep-Sea Research and Oceanographic Abstracts, 13, 699–705, 1966.
Morse, J. W. and Mackenzie, F. T.: Geochemistry of sedimentary carbonates, Elsevier, ISBN 9780444887818, 1990.
Morse, J. W., Arvidson, R. S., and Lüttge, A.: Calcium carbonate formation and dissolution, Chem. Rev., 107, 342–381, 2007.
Mucci, A.: The solubility of calcite and aragonite in seawater at various salinities, temperatures, and one atmosphere total pressure, Am. J. Sci., 283, 780–799, 1983.
Mucci, A., Sundby, B., Gehlen, M., Arakaki, T., Zhong, S., and Silverberg, N.: The fate of carbon in continental shelf sediments of eastern Canada: a case study, Deep-Sea Res. Pt. II, 47, 733–760, 2000.
Myers, T. J.: Buffering Estuarine Sediments Against Acidification, Master's Thesis, 77 pp., Oregon State University, https://ir.library.oregonstate.edu/concern/graduate_thesis_or_dissertations/br86bb964 (last access: 13 January 2024), 2022.
Pacella, S. R., Brown, C. A., Waldbusser, G. G., Labiosa, R. G., and Hales, B.: Seagrass habitat metabolism increases short-term extremes and long-term offset of CO2 under future ocean acidification, P. Natl. Acad. Scie. USA, 115, 3870–3875, 2018.
Pacella, S. R., Brown, C. A., Labiosa, R. G., Hales, B., Mochon Collura, T. C., Evans, W., and Waldbusser, G. G.: Feedbacks between estuarine metabolism and anthropogenic CO2 accelerate local rates of ocean acidification and hasten threshold exceedances, J. Geophys. Res.-Oceans, 129, e2023JC020313, https://doi.org/10.1029/2023JC020313, 2024.
Paul, A. J., Haunost, M., Goldenberg, S. U., Hartmann, J., Sánchez, N., Schneider, J., Suitner, N., and Riebesell, U.: Ocean alkalinity enhancement in an open ocean ecosystem: Biogeochemical responses and carbon storage durability, EGUsphere [preprint], https://doi.org/10.5194/egusphere-2024-417, 2024.
Rao, A. M., Malkin, S. Y., Montserrat, F., and Meysman, F. J.: Alkalinity production in intertidal sands intensified by lugworm bioirrigation. Estuarine, Coast. Shelf Sci., 148, 36–47, 2014.
Rassmann, J., Lansard, B., Pozzato, L., and Rabouille, C.: Carbonate chemistry in sediment porewaters of the Rhône River delta driven by early diagenesis (northwestern Mediterranean), Biogeosciences, 13, 5379–5394, https://doi.org/10.5194/bg-13-5379-2016, 2016.
Renforth, P. and Henderson, G.: Assessing ocean alkalinity for carbon sequestration, Rev. Geophys., 55, 636–674, https://doi.org/10.1002/2016RG000533, 2017.
Riley, J. P.: The occurrence of anomalously high fluoride concentrations in the North Atlantic, Deep-Sea Res. Pt. I, 12, 219–220, https://doi.org/10.1016/0011-7471(65)90027-6, 1965.
Rooze, J., Egger, M., Tsandev, I., and Slomp, C. P.: Iron-dependent anaerobic oxidation of methane in coastal surface sediments: Potential controls and impact, Limnol. Oceanogr., 61, S267–S282, 2016.
Rooze, J., Peterson, L., Peterson, R. N., and Meile, C.: Porewater flow patterns in surficial cold seep sediments inferred from conservative tracer profiles and early diagenetic modeling, Chem. Geol., 536, 119468, https://doi.org/10.1016/j.chemgeo.2020.119468, 2020.
Santos, I. R., Eyre, B. D., and Huettel, M.: The driving forces of porewater and groundwater flow in permeable coastal sediments: A review, Estuar. Coast. Shelf Sci., 98, 1–15, 2012.
Soetaert, K. and Meysman, F.: Reactive transport in aquatic ecosystems: Rapid model prototyping in the open source software R, Environ. Model. Softw., 32, 49–60, 2012.
Soetaert, K., Herman, P. M., and Middelburg, J. J.: A model of early diagenetic processes from the shelf to abyssal depths, Geochim. Cosmochim. Ac., 60, 1019–1040, 1996.
Soetaert, K., Hofmann, A. F., Middelburg, J. J., Meysman, F. J., and Greenwood, J.: The effect of biogeochemical processes on pH, Mar. Chem., 106, 380–401, 2007.
Soetaert, K., Petzoldt, T., and Meysman, F.: Marelac: Tools for aquatic sciences, https://cran.r-project.org/package=marelac (last access: - 13 January 2024), 2010.
Subhas, A. V., Rollins, N. E., Berelson, W. M., Erez, J., Ziveri, P., Langer, G., and Adkins, J. F.: The dissolution behavior of biogenic calcites in seawater and a possible role for magnesium and organic carbon, Mar. Chem., 205, 100–112, 2018.
Taylor, L. L., Quirk, J., Thorley, R. M. S., Kharecha, P. A., Hansen, J., Ridgwell, A., Lomas, M. R., Banwart, S. A., and Beerling, D. J.: Enhanced weathering strategies for stabilizing climate and averting ocean acidification, Nat. Clim. Change, 6, 402–406, 2016.
Thamdrup, B. and Canfield, D. E.: Benthic respiration in aquatic sediments, in: Methods in Ecosystem Science, New York, NY, Springer New York, 86–103, https://doi.org/10.1007/978-1-4612-1224-9_7, 2000.
Thomas, H., Schiettecatte, L.-S., Suykens, K., Koné, Y. J. M., Shadwick, E. H., Prowe, A. E. F., Bozec, Y., de Baar, H. J. W., and Borges, A. V.: Enhanced ocean carbon storage from anaerobic alkalinity generation in coastal sediments, Biogeosciences, 6, 267–274, https://doi.org/10.5194/bg-6-267-2009, 2009.
Thullner, M., Dale, A. W., and Regnier, P.: Global-scale quantification of mineralization pathways in marine sediments: A reaction-transport modeling approach, Geochem. Geophy. Geosy., 10, Q10012, https://doi.org/10.1029/2009GC002484, 2009.
Uppström, L. R.: The boron/chlorinity ratio of deep-sea water from the Pacific Ocean, Deep-Sea Res. Pt. I, 21, 161–162, 1974.
Waldbusser, G. G. and Salisbury, J. E.: Ocean acidification in the coastal zone from an organism's perspective: multiple system parameters, frequency domains, and habitats, Annu. Rev. Mar. Sci., 6, 221–247, 2014.
Waldbusser, G. G., Steenson, R. A., and Green, M. A.: Oyster shell dissolution rates in estuarine waters: effects of pH and shell legacy, J. Shellfish Res., 30, 659–669, 2011.
Waldbusser, G. G., Powell, E. N., and Mann, R.: Ecosystem effects of shell aggregations and cycling in coastal waters: an example of Chesapeake Bay oyster reefs, Ecology, 94, 895–903, 2013.
Wang, Y. and Van Cappellen, P.: A multicomponent reactive transport model of early diagenesis: Application to redox cycling in coastal marine sediments, Geochim. Cosmochim. Ac., 60, 2993–3014, 1996.
Wolf-Gladrow, D. A., Zeeber, R. E., Klaas, C., Koertzinger, A., and Dickson, A. G.: Total alkalinity: The explicit conservative expression and its application to biogeochemical processes, Mar. Chem., 106, 287–300, 2007.
Zeebe, R. E. and Wolf-Gladrow, D: CO2 in seawater: equilibrium, kinetics, isotopes, No. 65, Gulf Professional Publishing, 2001.