the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Jan 2020
| 22 Jan 2020
Organic iron complexes enhance iron transport capacity along estuarine salinity gradients of Baltic estuaries
Simon David Herzog
Per Persson
Kristina Kvashnina
Emma Sofia Kritzberg
Rivers discharge a notable amount of dissolved Fe (1.5×109 mol yr−1) to coastal waters but are still not considered important sources of bioavailable Fe to open marine waters. The reason is that the vast majority of particular and dissolved riverine Fe is considered to be lost to the sediment due to aggregation during estuarine mixing. Recently, however, several studies demonstrated relatively high stability of riverine Fe to salinity-induced aggregation, and it has been proposed that organically complexed Fe (Fe-OM) can “survive” the salinity gradient, while Fe (oxy)hydroxides are more prone to aggregation and selectively removed. In this study, we directly identified, by X-ray absorption spectroscopy, the occurrence of these two Fe phases across eight boreal rivers draining into the Baltic Sea and confirmed a significant but variable contribution of Fe-OM in relation to Fe (oxy)hydroxides among river mouths. We further found that Fe-OM was more prevalent at high flow conditions in spring than at low flow conditions during autumn and that Fe-OM was more dominant upstream in a catchment than at the river mouth. The stability of Fe to increasing salinity, as assessed by artificial mixing experiments, correlated well to the relative contribution of Fe-OM, confirming that organic complexes promote Fe transport capacity. This study suggests that boreal rivers may provide significant amounts of potentially bioavailable Fe beyond the estuary, due to organic matter complexes.
- Article
(2936 KB) - Full-text XML
-
Supplement
(772 KB) - BibTeX
- EndNote





Iron (Fe) mobility from the lithosphere and pedosphere into the hydrosphere and biosphere is controlled by physical, chemical and biological processes. While Fe is the fourth most abundant element in the Earth's crust (Taylor, 1964), Fe concentrations in oxygenated aquatic systems are generally low (Johnson et al., 1997; Kraemer, 2004), but they can be higher during high flow conditions and in boreal waters with high dissolved organic carbon (DOC) concentrations (Kritzberg et al., 2014; Ekström et al., 2016). The more soluble form of Fe, i.e., Fe(II), is favored under strongly reducing or highly acidic conditions (Waychunas et al., 2005). At circumneutral pH and oxic conditions Fe(II) gets oxidized to Fe(III), which has a low solubility and precipitates as Fe (oxy)hydroxides (Lofts et al., 2008; Liu and Millero, 2002). Thus, mobilization of Fe to surface waters requires either water flow through anoxic soil layers, favoring Fe(II), or that Fe is complexed by organic ligands and also becomes mobile in oxic soil layers (Tipping, 1981; Stumm and Morgan, 1970).
Several studies report rising Fe concentrations in surface waters, especially in northern Europe (Neal et al., 2008; Kritzberg and Ekström, 2012; Sarkkola et al., 2013; Weyhenmeyer et al., 2014; Björnerås et al., 2017), suggesting that Fe export from soils is increasing. As a consequence, Fe loading from boreal rivers to estuaries is increasing substantially (Kritzberg and Ekström, 2012; Kritzberg et al., 2014; Björnerås et al., 2017). Given the key role that Fe plays in both local and global biogeochemical cycles in coastal and marine systems, this is a finding with major implications for the receiving systems. What the consequences may be, depends first and foremost on the fate of Fe in the estuarine salinity gradient. To the extent that Fe is stable to salinity-induced aggregation and sedimentation, it may provide potentially bioavailable Fe to the marine system. However, Fe is known to behave nonconservatively in estuaries, and it has been suggested that at least 95 % of Fe is aggregated and lost to the sediments in the early part of estuarine mixing (Sholkovitz et al., 1978; Haese, 2006). Fe can play an important role in the sediment, e.g., by acting as a C and P sink (Lalonde et al., 2012; Lenstra et al., 2018). Moreover, benthic release of Fe and subsequent lateral transport was recently found to be a significant source of dissolved Fe to open marine waters (van Hulten et al., 2017).
Fe transport capacity – the fraction of riverine Fe remaining in suspension at higher salinity – has been shown to vary widely and is in some instances much higher than predicted from prior work (Kritzberg et al., 2014; Krachler et al., 2005). Thus, the riverine Fe source to marine waters may be underestimated, especially for boreal rivers, where high DOC concentrations can affect Fe speciation. Fe in natural waters is known to occur in two main phases, mononuclear organic complexes (Fe-OM) and Fe-rich Fe (oxy)hydroxide colloids associated with organic matter (Breitbarth et al., 2010; Hassellöv et al., 1999; Andersson et al., 2006). It has been suggested that variability in Fe transport capacity between rivers (1 % to 55 %) may be explained by the varying proportion of these Fe phases (Kritzberg et al., 2014). Further, studies using cathodic stripping voltammetry (CSV) have underlined the importance of complexation by ligands for keeping Fe in suspension in saline waters (Laglera and van den Berg, 2009; Sukekava et al., 2018). However, the postulated link between Fe-OM and Fe transport capacity requires direct assessment of Fe speciation to verify previous interpretations based on Fe : OC ratios and molecular size (Stolpe and Hassellöv, 2007; Krachler et al., 2010; Kritzberg et al., 2014). A preferential loss of Fe (oxy)hydroxides by aggregation was shown by Herzog et al. (2017) using X-ray absorbance spectroscopy (XAS). While this infers that Fe-OM is more stable to salinity-induced aggregation, some Fe-OM was also found in aggregates, indicating that the control of Fe stability is more complex.
Based on the previous findings, the aim of the current study was to better understand what controls the fate of Fe from boreal rivers across estuarine salinity gradients by exploring (1) if variability in relative contribution of Fe-OM and Fe (oxy)hydroxides can explain variation in Fe transport capacity and (2) if the relative contribution of Fe-OM and Fe (oxy)hydroxide is controlled by spatial factors and flow conditions, within and among rivers.
To this purpose, we sampled eight river mouths that drain at the Swedish coast into the brackish Baltic Sea. The rivers were chosen to encompass a wide geographical and climatic gradient, as well as a range in Fe and DOC concentrations. The Fe speciation, organic speciation (organic vs. inorganic) and redox speciation were characterized by XAS. To be able to link Fe speciation to Fe transport capacity, the same river waters were exposed to artificial salinity gradients (mixing experiments). Four of the rivers were sampled under high (spring) and low flow conditions (autumn). To reveal differences along the flow path, two upstream sites were sampled in addition to the river mouth in one river catchment.
2.1 Site description and sampling
Eight rivers around the Swedish coast, with distinct differences in climate and catchment characteristics (Fig. 1), were selected for this study. The annual temperature ranged from 5.8 ∘C for the most northern (Öre) and 9.8 ∘C for the most southern river mouth (Helge). Forest is the most dominant land cover and peat soils are present to a varying extent in all the catchments (Table 1). While six of these river mouths were sampled for previous studies exploring Fe dynamics in response to increasing salinity (Kritzberg et al., 2014; Herzog et al., 2017), relating the XAS-assessed contribution of Fe-OM complexes to Fe transport capacity, was not previously done. Moreover, to investigate differences in Fe speciation along a river, two upstream locations in Helge river were included. The most upstream sample site, Svineö, which drains from a peat bog, has a high percentage of peat soil. The second site along the river path, Biveröd, is a small second-order stream in a predominantly forested landscape. In addition, to understand the impact of high and low flow conditions on Fe speciation and transport capacity, sampling was carried out twice – during autumn and spring – in four of the river mouths (Emån, Lyckby, Mörrum and Helge). A major difference in the discharge between the autumn and spring sampling was observed (Table 1). Finally, for two rivers (Öre and Örekil), transects were sampled starting at the river mouth and extending over the estuarine salinity gradient, to facilitate comparison of Fe transport capacity by mixing experiments and in situ Fe concentrations along the natural salinity gradient.
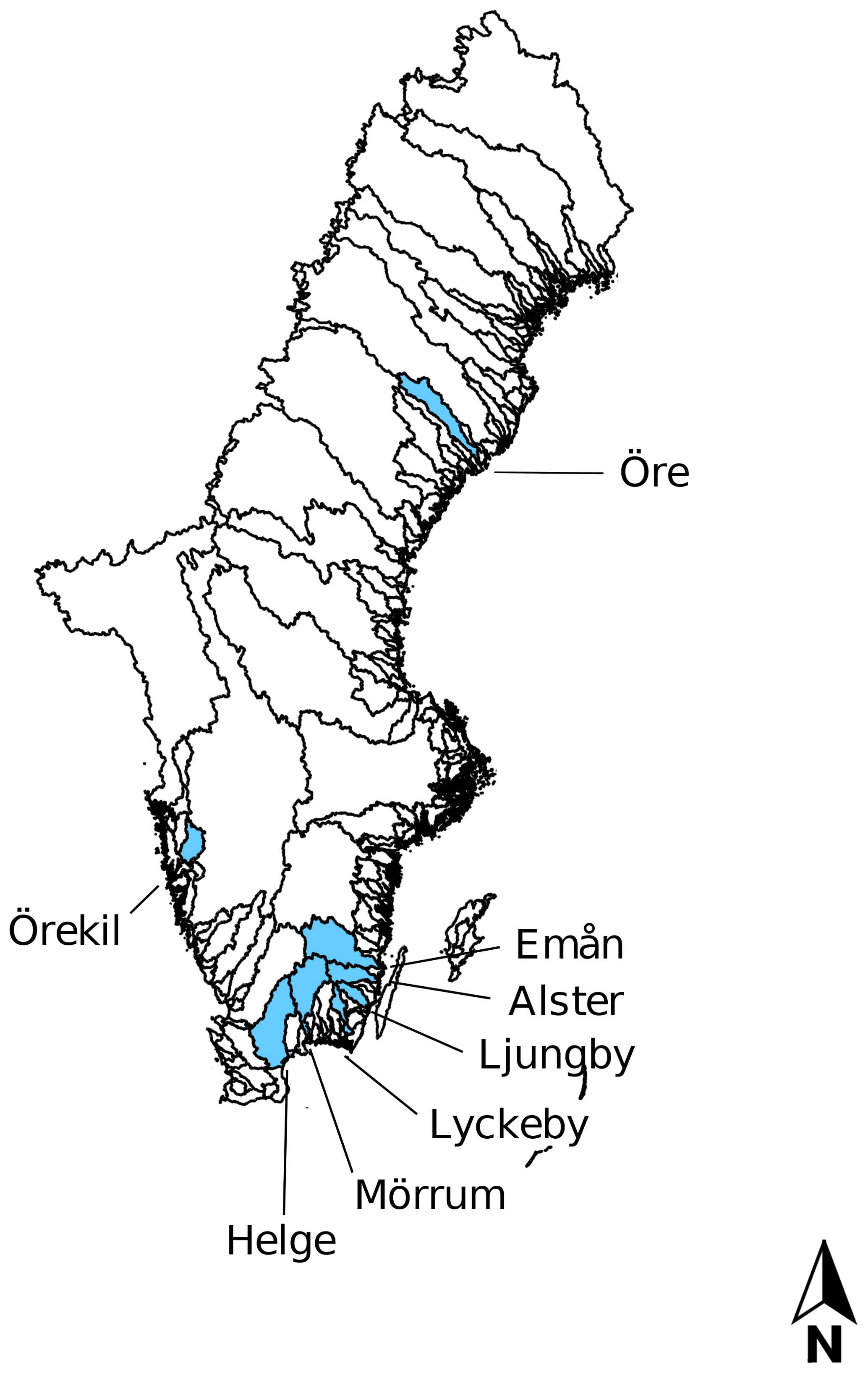
Figure 1Map of all river catchments in Sweden with the ones considered in this study named and marked blue.
Table 1Catchment characteristics.
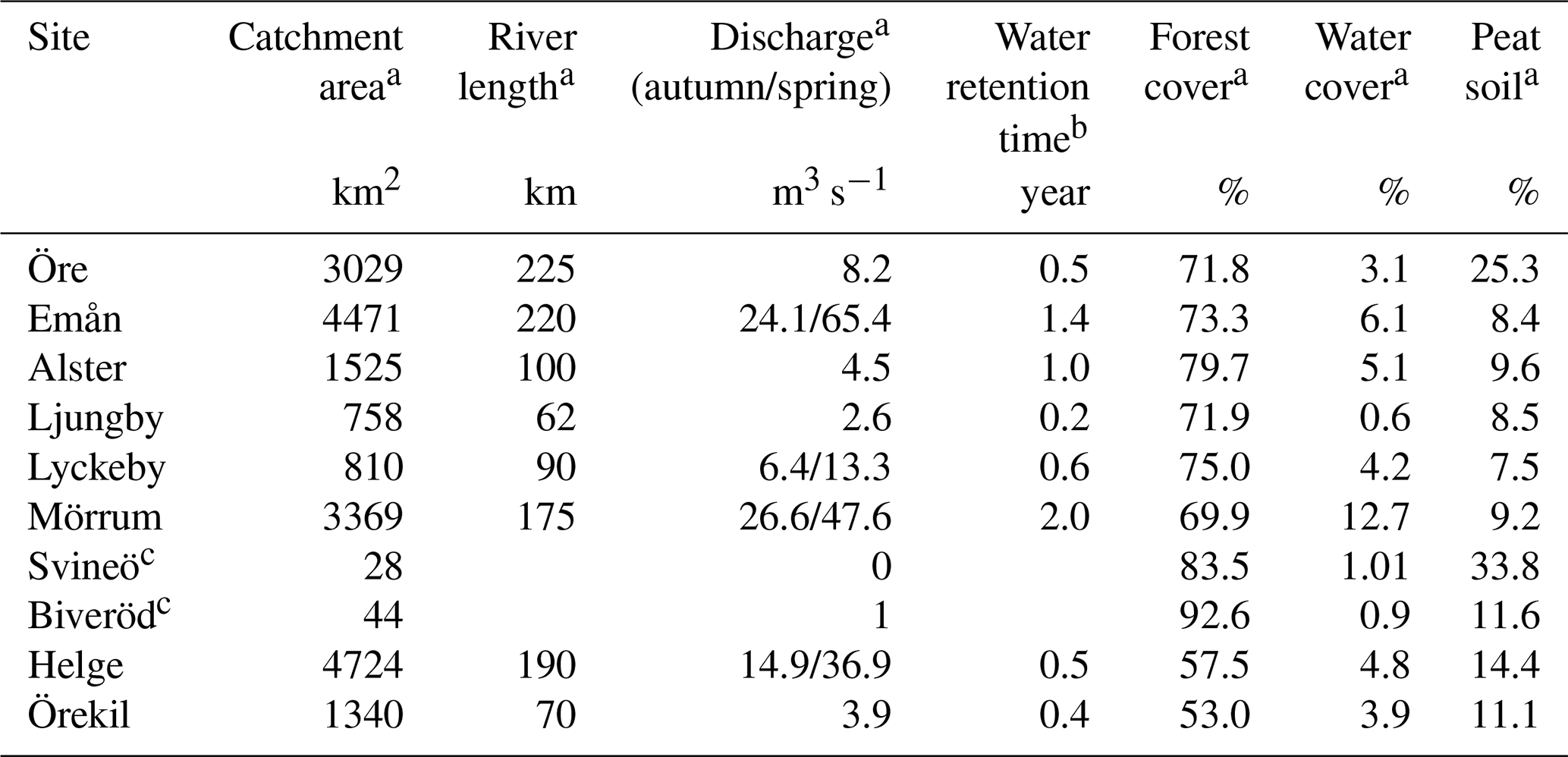
a Data obtained from http://vattenweb.smhi.se (last access: 23 June 2018). b Data from Lindström et al. (2018). c Upstream sites in the Helge catchment.
Water was sampled by hand from half a meter below the surface into acid-cleaned polyethylene containers through a 150 µm nylon mesh. The mesh was used to ensure homogeneous samples free of large detritus. No further filtration steps were applied, to ensure that all suspended Fe was included, which is critical when studying the stability of riverine Fe across salinity gradients. Samples were stored cold and dark in a cooling box with freezing elements until they are returned to the lab.
For the XAS analysis, a 1 L sample of water was frozen as soon as possible and never more than 5 h after collection. The samples were later freeze-dried and stored dry in the dark until analyzed. Freeze-drying is commonly applied for preservation and pre-concentration of XAS samples (Karlsson et al., 2008; Vilgé-Ritter et al., 1999). While the freezing may lead to changes in the physical properties of colloids (Raiswell et al., 2010), the chemical composition should be less affected. For instance, drying or freeze-drying could increase the rate of crystallization of Fe (Bordas and Bourg, 1998), reducing the amorphous Fe phase compared with native samples, but this would not affect the distinction between organically complexed Fe and Fe (oxy)hydroxides, which was the focus here.
Oxygen and pH were measured in situ with OxyGuard MkIII and SevenGo Duo pH meter (Mettler Toledo), respectively. Only acid-washed material was used for sample handling, and for XAS and total Fe measurement polycarbonate bottles and containers were used.
2.2 Artificial seawater mixing experiments
Experiments mixing river water and artificial seawater were initiated as soon as possible and no later than 3 h after sampling. Water samples were mixed with artificial sea salt solutions in a 6 : 1 ratio (vol : vol) in 50 mL Falcon tubes to 8 or 16 levels of salinity ranging from 0 to 35. To achieve the desired final salinities the added concentration of the sea salt solution varied. These were made from an artificial sea salt stock solution produced using reagent grade salts (Sigma Aldrich) following a standard protocol (Kester et al., 1967) (mass fraction given in %: Cl− (55.05), Na+ (30.62), (7.68), Mg2+ (3.69), Ca2+ (1.15), K+ (1.10), (0.40), Br− (0.19), H3BO3 (0.07), Sr2+ (0.04), F− (0.003)). The Fe contamination from the salts used was negligible, e.g., the addition of salt to produce salinity 35 added a maximum of 0.0025 mg L−1 Fe, which corresponds to 0.1 %–3.8 % of the Fe concentration in the river waters studied. The stock solution was diluted to the desired concentration by Milli-Q water (Millipore, 18.2 MΩ). After mixing the river water with the salt solution, the samples were kept in the dark on a shaker for at least 24 h to allow aggregation. Salinity-induced aggregation of Fe consists of several reactions with a significant fraction aggregating within a few seconds (Nowostawska et al., 2008). While aggregation then continues at a slower rate after the first few hours (Nowostawska et al., 2008; Hunter and Leonard, 1988), the first 24 h should include the largest fraction of the Fe removal.
The aggregates were separated by centrifugation at 3000 g for 8 h at 4 ∘C. After centrifugation, Fe, DOC, pH and salinity were measured in the supernatant. Fe transport capacity was calculated as the Fe concentration in the supernatant divided by the in situ Fe concentration and multiplied by 100 (%).
2.3 Standard analytical methods
An ICP-AES Optima 3000DV (Perkin Elmer) was used to determine total Fe concentration on acidified samples (1 % vol, HNO3). A Shimadzu TOC V-CPN was used to analyze organic carbon by high-temperature catalytic-oxidation, using the non-purgeable organic carbon (NPOC) method. For calibration, a five-point standard curve was used and blanks and standards were included in all runs. The pH of the mixing experiment samples was measured with a 913 pH Meter (Metrohm) and salinity was determined by a WTW inoLab Cond730.
2.4 XAS data collection and analysis
Synchrotron data were collected at the beam line I811 MaxII ring (Max Lab, Lund University). Fe K-edge XAS spectra were collected on the river samples at fluorescence mode at room temperature. A Lytle detector with an Mn filter (3 µx) was used to minimize unwanted scattering and fluorescence contributions. Aligned samples at 45∘ relative to the incident beam guaranteed an optimal fluorescence signal. Depending on the Fe concentration 15 to 40 spectra for each samples were recorded taking approximately 5 min each. Transmission scans of a reference Fe foil were collected simultaneously during all scans for energy calibration.
Data treatment and analysis for the extended X-ray absorption fine structure (EXAFS) and wavelet transform (WT) spectra was performed according to Herzog et al. (2017). A subset of the samples (6 out of 14) was previously analyzed for EXAFS (Herzog et al., 2017), and reanalysis was performed with consistent fitting parameters for the whole data set to allow comparison among all samples. In short, all scans were checked for beam damage before being averaged with SixPack (Webb, 2005). The averaged scans were normalized and the background was removed by subtracting a spline function in Viper (Klementiev, 2000). The same program was used for investigation of self-absorbance and shell by shell fitting of the EXAFS data. For the WT analysis Igor Pro script was used (Funke et al., 2005). The k3-weighted spectra were modeled in k-space from 2.8 to 12.0 Å−1 using theoretical phase and amplitude functions from FEFF7 (Zabinsky et al., 1995). Goethite (O'day et al., 2004) and the trisoxalatoiron(III) complex (Persson and Axe, 2005) were used as input structures for calculations with FEFF. While fitting, the amplitude reduction factor () was set to 0.70. Further, with values found in the literature, the numbers of free variables were restricted by correlating coordination numbers and fixing the Debye–Waller factors (σ2). Also, the threshold energy (ΔE0) beyond the first was assumed to be identical for all shells.
A linear combination fitting (LCF) analysis was applied to the river mouth samples using SixPack. k3-weighted EXAFS spectra (k3.0 to 12.0 Å−1) were used in the LCF analysis. Reference spectra of ferrihydrite, goethite, hematite, lepidocrocite and a Fe(III) complexed to Suwannee Rives fulvic acid were used as model compounds. This model provided good fits for all samples and further allowed us to distinguish between organically complexed Fe and Fe (oxy)hydroxide. During the LCF analysis E0 was allowed to float, a nonnegative boundary condition was applied and the sum of species was not forced to equal 100 %. Components with a contribution less than 5 % were excluded from the models.
Fe K-edge high-energy-resolution fluorescence detection (HERFD) XANES spectra and Fe Kβ2,5 emission spectra were collected at the high-brilliance X-ray absorption and X-ray emission spectroscopy undulator beamline ID26 of the European Synchrotron Radiation Facility (ESRF, Grenoble) (Signorato et al., 1999). The incident X-ray beam was monochromatized with a pair of cryogenically cooled Si(111) crystals. The sample, analyzer crystal and photon detector (silicon drift diode) were arranged in a vertical Rowland geometry. The intensity was normalized to the incident flux. Both the HERFD and the Fe Kβ2,5 emission spectra were measured using the X-ray emission spectrometer (Glatzel and Bergmann, 2005; Kvashnina and Scheinost, 2016), and the HERFD measurements were performed by recording the intensity of the Fe Kβ1 emission line (7058 eV) as a function of the incident energy using five Ge (620) crystal analyzers at 79∘ Bragg angle.
2.5 Data treatment
The contribution of the single Fe-C and the multiple Fe-O∕C scattering paths was statistically analyzed in the EXAFS fits by an F test with Viper (Klementev, 2001).
As a measure of the relative contribution of Fe-OM and Fe (oxy)hydroxides in the water samples, two different approaches using the k3-weighted EXAFS spectra were applied: (1) a ratio of the coordination numbers of the fitting results, between the Fe-C path and the shortest (edge-sharing) Fe-Fe path (i.e., CNFe-C∕CNFe-Fe) and (2) a ratio of the Fe-OM fraction and the sum of Fe-oxide fractions from the LCF analysis.
Relationships in the river mouth samples between Fe transport capacity at 35 salinity (corresponding to the salinity of the open sea), CNFe-C∕CNFe-Fe ratios, LCF ratio, Fe : OC ratios, total Fe and DOC were tested by Pearson correlations. Assumptions of normality were verified by Shapiro–Wilk tests. Differences in Fe transport capacity between spring and autumn samples, as well as DOC concentrations at in situ and 35 salinity in the mixing experiment, were tested by paired t tests.
Expected, or theoretical, values of in situ Fe across the estuarine salinity gradient were calculated by accounting for the dilution of riverine water by seawater (estimated by salinity) and the stability of Fe to aggregation as assessed by the artificial mixing experiments. The following equation Eq. (1) was used to calculate the expected Fe concentration [Feexp] at a given salinity:
where [Feriver] is the Fe concentration at the river mouth, salinitymarine end-member is the highest salinity in the estuarine in situ gradient, [Femarine end-member] is the Fe concentration at the highest salinity in the estuarine gradient and Festability is the fraction that remained in suspension in the artificial seawater mixing experiments at the given salinity.
3.1 Water chemistry
At the time of sampling all river mouths were close to saturation with dissolved oxygen (85 %–118 %) and pH values close to neutral (Table 2). For the river mouths that were sampled twice, pH was consistently lower during spring than during autumn sampling, i.e., lower during higher discharge. Total Fe concentrations in the river mouths varied from 0.22 to 2.28 mg L−1 and DOC concentrations from 8.8 to 24.2 mg L−1. Water chemistry in the two upstream samples from the Helge catchment differed strongly from those of the river mouth. Oxygen saturation and pH were lower in the upstream sites, especially in Svineö (dissolved oxygen saturation 41 % and pH 4.4), while Fe and DOC concentrations were markedly higher. Across all samples Fe and DOC concentrations were strongly correlated (r=0.96; p<0.001), but since Fe was more variable (32-fold) than DOC (6-fold), there was a wide range in Fe : DOC molar ratios, from 0.005 to 0.035.
Table 2Water chemistry and transport capacity at salinity 35 (corresponding to the salinity of the open sea) of collected samples.

aRatio of the coordination numbers of the fitting results, between the Fe-C path and the shortest (edge-sharing) Fe-Fe path (i.e., CNFe-C∕CNFe-Fe). b Ratio of the Fe-OM fraction and the sum of Fe-oxide fractions from the LCF analysis. c Upstream sites in the Helge catchment.
3.2 XAS characterization
The XAS analyses identified two main Fe phases, namely Fe (oxy)hydroxide, and Fe ions associated with organic matter as Fe-OM complexes. Both phases were qualitatively identified in the WT contour plots (Fig. 2). The feature in the WT plots at ca. 7.5 Å−1, 2.8 Å, originate from Fe-Fe scattering paths (denoted Fig. 2a as Fe) and are similar to that of ferrihydrite (Fig. S2 in the Supplement) (Sundman et al., 2014; Yu et al., 2015) and goethite (Fig. S2) (Karlsson and Persson, 2010; Sundman et al., 2014). Features for the Fe-OM complexes occurred at ca. 3 Å−1, 2.5 Å and 3 Å−1, 3.2–3.7 Å, caused by single Fe-C and multiple Fe-C-C(O or N) scattering, respectively (denoted in Fig. 2a as C and C∕O), and are in good agreement with previously identified Fe-OM complexes (Fig. S2) (Karlsson and Persson, 2010). In Fig. 2 only a selection of the WT plots are shown; all WT plots of the remaining samples can be found in Fig. S1.
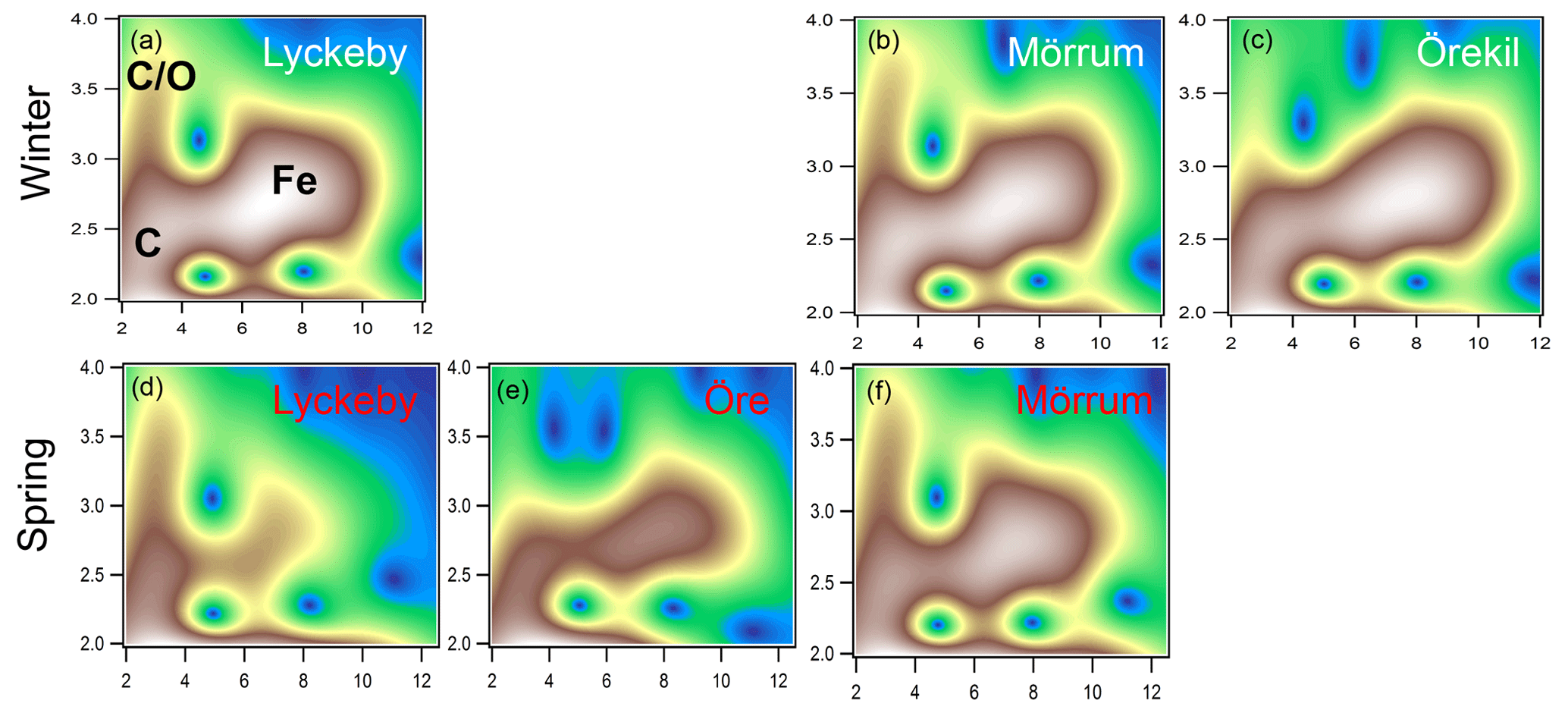
Figure 2Morlet wavelet transforms (η=4, σ=2) of EXAFS data collected on samples from the Lyckeby, Mörrum, Örekil and Öre rivers (white represents autumn and red represents spring) and are plotted as a function of k (Å−1) on the x axis and R (Å) on the y axis. In panel (a) (Lyckeby) areas representing the different Fe scattering paths are indicated by C (Fe-C), C∕O (Fe-C-C∕O) and Fe (Fe-Fe).
Guided by the WT results, the EXAFS spectra were quantitatively modeled by a shell-by-shell nonlinear least-squares fitting procedure (Table S2 in the Supplement) including five paths (Fe-O, two Fe-Fe, Fe-C and a Fe-C∕O multiple-scattering path). The two Fe-Fe paths were used to describe the contribution of Fe (oxy)hydroxide and the Fe-C, and the Fe-C∕O multiple-scattering paths were used for the Fe-OM component. This modeling approach corroborated the qualitative WT analyses and provided good fits to all spectra (Fig. S3). For the river mouth samples the Fe-Fe edge- and corner-sharing distances were determined to 3.05–3.11 and 3.41–3.46 Å, respectively (Table S1). The coordination number (CN) of the short Fe-Fe path varied between 1.0 and 2.7, indicating significant contribution from Fe (oxy)hydroxide, as corroborated by the WT plots. The Fe-C distances varied between 2.85 and 3.00 Å, and the CN of the Fe-C path varied between 0.9 and 2.8 Å. To verify the contribution of the Fe-C path, an F test comparing EXAFS models with and without the Fe-C and the Fe-C∕O multiple-scattering paths was performed, showing a significant contribution at the 92 % confidence level or better. While the EXAFS-fitting analyses confirmed the presence of both Fe (oxy)hydroxide and Fe-OM complexes in all river mouth samples, the CN values indicated a large variation in the relative contribution of these Fe species (Table S1).
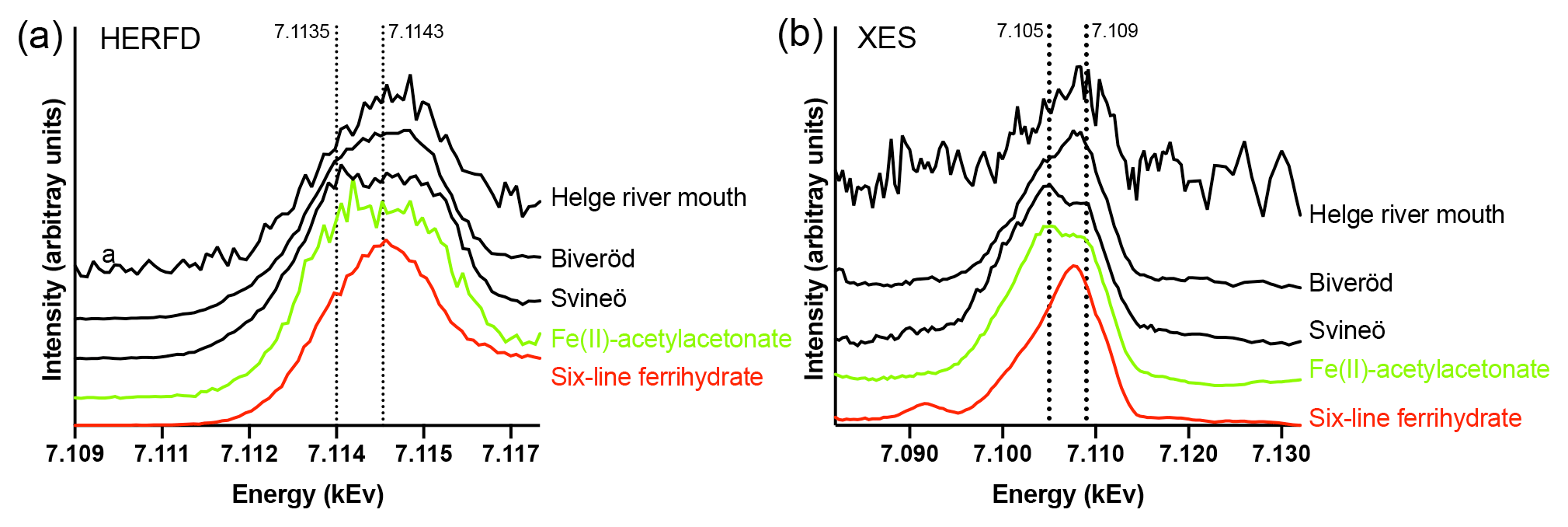
Figure 3Fe K-edge HERFD pre-edge spectra (a) and Kβ2,5 X-ray emission (XES) spectra (b) of the Helge river mouth sample, the Biveröd sample, the Svineö sample, Fe(II)-acetylacetonate (Fe(acac)2) (green), and six-line Ferrihydrite (red). The spectra were normalized to the maximum intensities. The dotted lines are included for visual guidance.
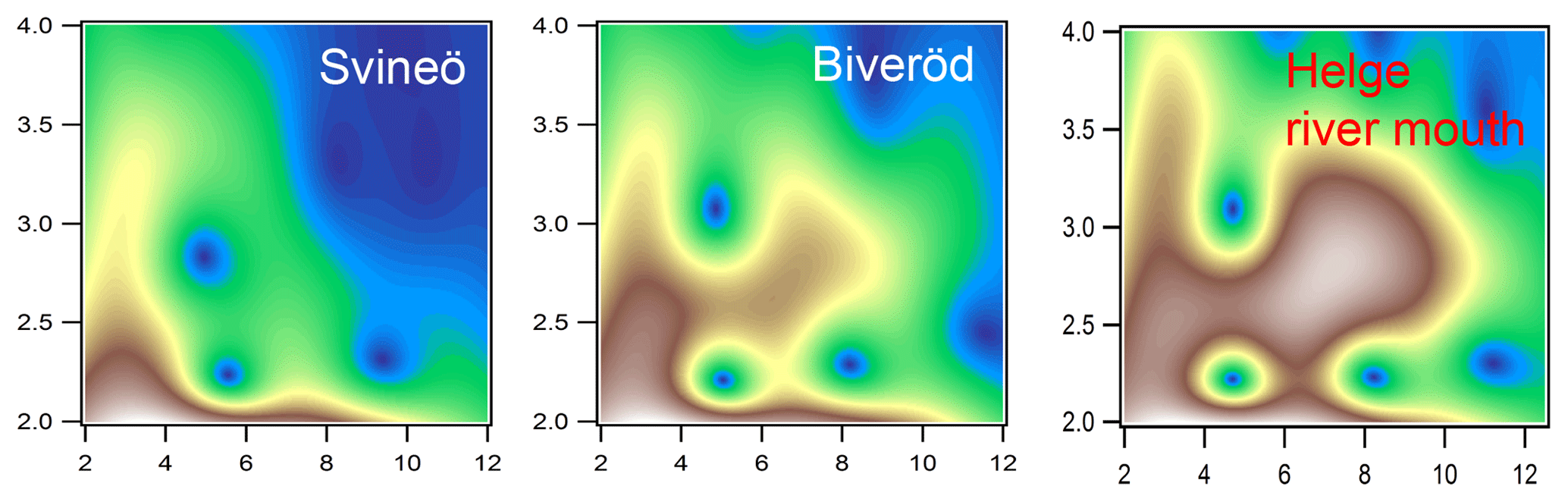
Figure 4Morlet wavelet transforms (η=4; σ=2) of EXAFS data of the two upstream samples from Svineö and Biveröd and the river mouth of the Helge catchment (white represents autumn and red represents spring), plotted as a function of k (Å−1) on the x axis and R (Å) on the y axis.
LCF analysis supported the variable contribution of the two Fe phases in the water samples (Table S2) and was in good agreement with the WT data. LCF analysis assigned the main components in the river mouth samples to Fe-OM, ferrihydrite and lepidocrocite. The contributions from goethite and hematite were below 5 % in all samples and therefore excluded from the final analysis.
The XAS data contain information on the local structure around the selected element (Fe) that it is not strictly quantitative. Therefore, the ratios were merely used to identify trends in the relative contribution of Fe-OM vs. Fe (oxy)hydroxides among the samples. Differences in the relative contribution of Fe-OM and Fe (oxy)hydroxide across the samples were obvious from the WT plots (Figs. 2 and S1) and supported by the CNFe-C∕CNFe-Fe and LCF ratios (Table 2). There was agreement between the two ratios, as indicated by correlations across all samples (r=0.58; p=0.047) and river mouth samples only (r=0.64; p=0.033) (Fig. S4). Low ratios, i.e., low contributions of Fe-OM, were indicated by both approaches for the Öre, Alster, Lyckebyautumn and Örekil samples, for which the Fe (oxy)hydroxide feature was dominant in the WT plots. This was contrasted by Lyckebyspring and Mörrumspring samples, with high ratios and particularly strong Fe-OM signals in the WT plots. For the other river mouth samples, a more even contribution of the two Fe species was found.
Fe speciation in the two samples taken upstream in the Helge river catchment was distinctly different from the river mouth. The HERFD and Kβ2,5 emission spectra (Fig. 3a and b) indicated a gradual change from low to high Fe oxidation state when approaching the river mouth. Due to low Fe concentration there was more noise in the river mouth sample, but the deviation from the Fe(II) spectra was still clear. In the HERFD spectra the presence of Fe(II) was shown by a low-energy shoulder at 7.1135 kEv, which was most pronounced for the Svineö sample (Fig. 3b). This effect was even more obvious in the Kβ2,5 emission spectra (Fig. 3a) where the peak at 7.105 kEv provided direct evidence for the presence of Fe(II), based on comparison with the model compounds. The emission spectrum of the Svineö sample is very similar to the organic Fe(II) complex Fe(acac)2, which suggests that Fe(II) was the predominant oxidation state at this site.
The WT results of the Svineö sample indicated no Fe-Fe scattering (Fig. 4), whereas further downstream at Biveröd both Fe-C and Fe-C-C(O or N) features were present. In the river mouth Fe-Fe was even further pronounced. Thus, the Fe-Fe signal increased in strength the further downstream the sample was taken.
The LCF analysis corroborated the WT results; i.e., for Svineö no Fe-oxides were identified, whereas in Biveröd both Fe-OM and Fe-oxides were present and in the Helge river mouth Fe-oxides were dominating (Table S2). This trend was also obvious from the quantitative shell-by-shell fitting results (Table S1). Finally, comparing the various EXAFS analyses with the HERFED and Kβ2,5 emission spectroscopy results show that Fe(II) in the upstream samples of the Helge river system is present as Fe-OM complexes. These complexes are favored by low pH and low oxygen concentrations, as expected.
For the river mouths that were sampled twice, samples taken during high flow regime in spring displayed consistently higher CNFe-C∕CNFe-Fe and LCF ratios than samples taken in the same river in autumn during lower discharge, indicating a higher contribution of Fe-OM complexes during high flow conditions (Table S1).
Considering all samples, the relative contribution of Fe-OM and Fe (oxy)hydroxide was not predicted by the molar Fe : OC, i.e., no correlation between the Fe : OC and the Fe speciation ratios was found (CNFe-C∕CNFe-Fe ratio: r=0.182, p=0.570; LCF ratio: r=0.53; p=0.077). Instead, the LCF ratio was negatively correlated to pH (; p=0.019). Among all samples, no significant relationships were found between CNFe-C∕CNFe-Fe or LCF ratios and variables related to catchment size, land cover or soil type.
3.3 Fe transport capacity
The general pattern of the artificial seawater mixing experiments was a nonconservative behavior of Fe with increasing salinity (Fig. 5). Fe removal already took place at low salinities, with more than 50 % of the Fe removed at salinity 2 for some river samples (Öre, Alster, Mörrumautumn and Helgespring, Helgeautumn). At a salinity corresponding to the open ocean (35), between 76 % and 93 % of Fe was removed. High Fe transport capacity was measured for Lyckeby river, with 24 % remaining in suspension at salinity 35. No significant loss of OC in response to increasing salinity was found for rivers where OC was analyzed in the mixing experiment (Öre, Örekil, Helge, Mörrum, Emån and Lyckeby: t5=1.38; p=0.17).
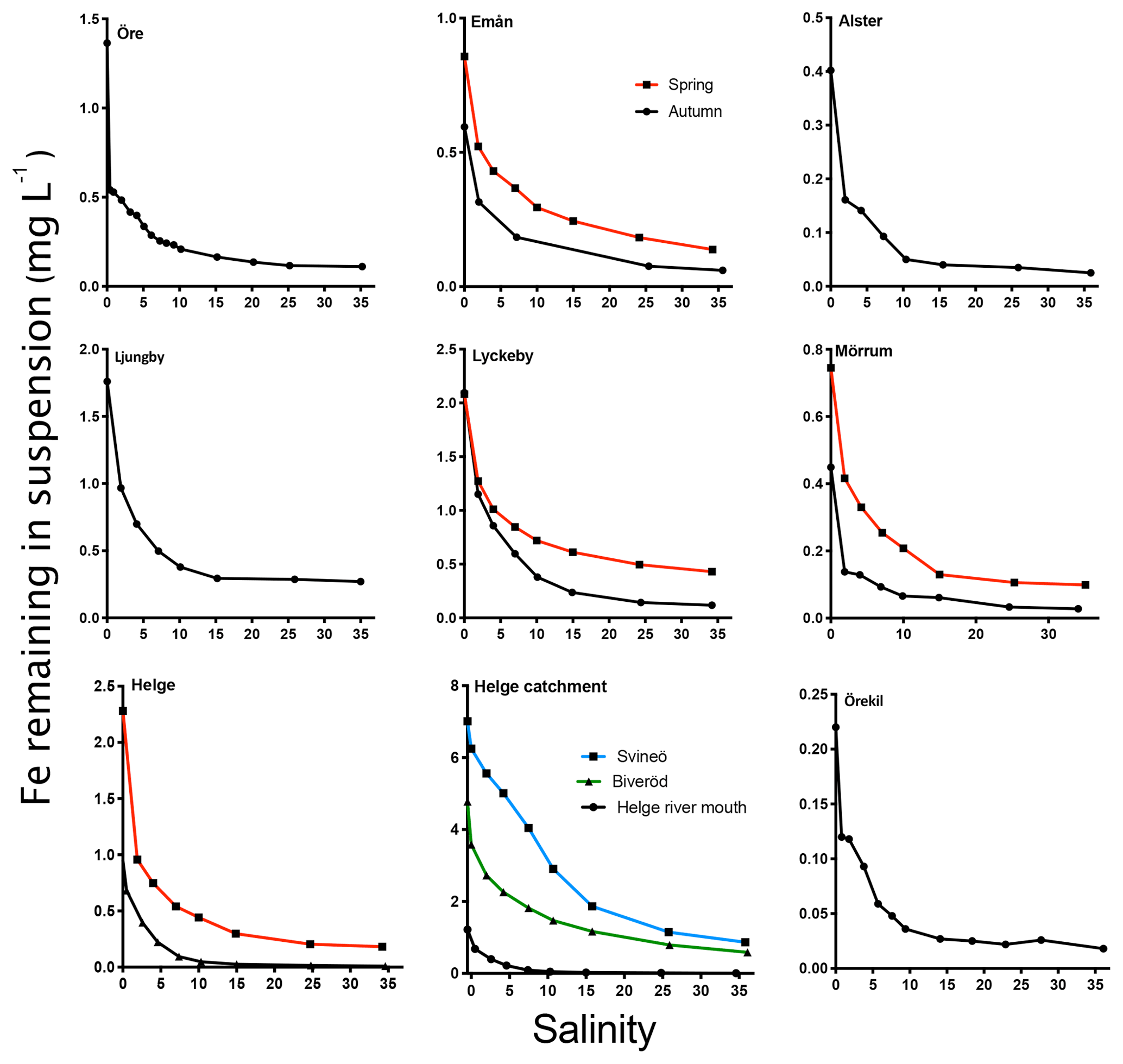
Figure 5The change in Fe in suspension in response to increasing salinity (0–35). Black lines denote sampling in autumn and red lines denote sampling in spring. For Helge river, the blue line denotes the most upstream sample (Svineö), the green line the other upstream sample (Biveröd) and the black line the river mouth.
For the river mouth samples, the Fe transport capacity at 35 salinity correlated positively with the Fe speciation ratios (CNFe-C∕CNFe-Fe: r=0.675; p=0.023; LCF ratio =0.78; p=0.005) (Fig. S5). Further, Fe transport capacity at 35 salinity was negatively correlated to pH (, p=0.007).
When comparing the samples taken within the Helge catchment, upstream samples showed a higher Fe transport capacity than the river mouth sample (Fig. 5). For the four river mouths that were sampled twice, Fe transport capacity (at 35) was consistently higher in spring than in autumn (t3=4.696, p=0.0183).
In addition to raising the salinity, the pH in the samples also increased due to the experimental treatments from 6.4 to 7.5 in the river mouths to 7.4–8.3 in the high-salinity treatments and from pH 4.7–6.5 to pH 7.1–7.7 for the upstream samples.
In situ concentrations of total Fe declined with increasing salinity and distance from the river mouths (Fig. 6). The theoretical values for Fe, calculated based on salinity-induced aggregation in the artificial seawater mixing experiments and dilution estimated by salinity, were only slightly lower than the measured in situ values for Örekil river. The deviation was larger for Öre river, where the Fe measured in situ was substantially higher than the theoretical values at the lower levels of salinity.
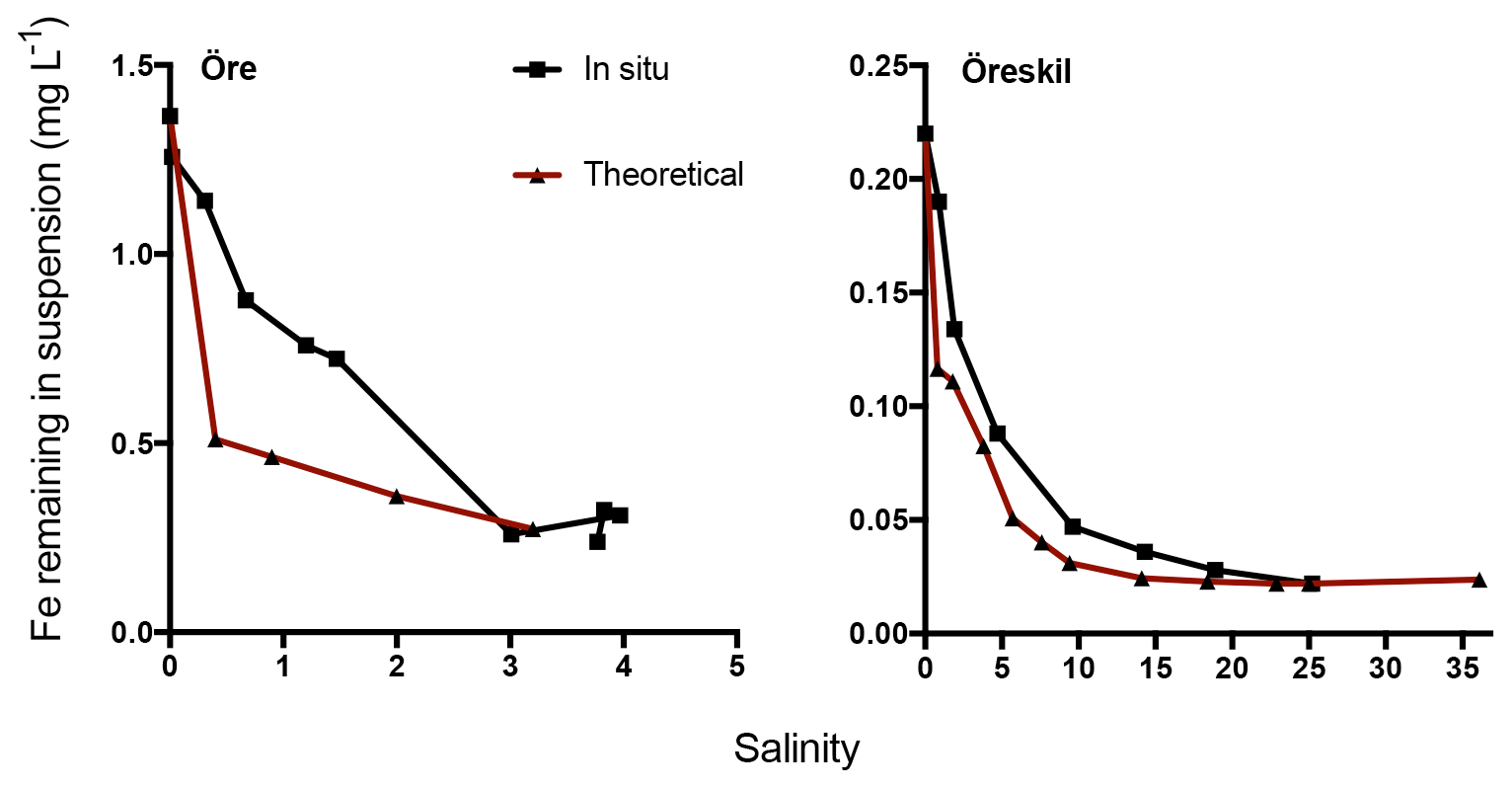
Figure 6Fe concentration in suspension at different salinities. The black line denotes in situ samples along a transect from the river mouth and the brown line theoretical values based on mixing experiments.
4.1 In situ speciation of Fe
Two carrier phases associated with Fe transport in freshwater in a boreal system – Fe-OM and Fe (oxy)hydroxides (Andersson et al., 2006; Hassellöv et al., 1999) – have been previously verified by XAS (Sundman et al., 2013; Herzog et al., 2017) and were also identified and detected in all river mouth samples in this study. The results of the quantitative modeling of the EXAFS spectra and LCF analysis correlated, which is satisfying considering the potential sources of error of both analyses. Results showed a wide variation in the relative contribution of the Fe phases across river mouths, with some dominated by Fe-OM, e.g., Lyckebyspring river samples, and some by Fe (oxy)hydroxide, e.g., Öre river. The significant contribution of Fe-OM in the river mouth samples is in contrast to the thermodynamic modeling suggested by Wällstedt et al. (2010) for these systems that predicted a dominance of ferrihydrite (97 %).
The contribution of the Fe phases changed markedly along the Helge river catchment. In the water draining from a peat bog (Svineö), Fe(II) was predominant and only Fe-OM was present. Neither EXAFS-fitting nor LCF identified Fe-Fe paths to support the presence of Fe (oxy)hydroxide. The sample showed similar features to groundwater in a northern boreal catchment (Sundman et al., 2014) or Fe(II) loaded onto peat humic acid (Yu et al., 2015). Further downstream (Biveröd), a mixture of the two phases could be seen, similar to samples from soil waters from organic layers close to a boreal stream (Sundman et al., 2014). The Fe(III) and Fe (oxy)hydroxide fraction was more prominent along the river path and was highest at the river mouth.
In addition to variation across river mouths and within the Helge river catchment, XAS analyses revealed a clear variation between high and low flow regime in the river mouth samples. Samples collected during high flow in spring showed a higher contribution of Fe-OM than the autumn samples, which is a direct support for previous interpretations made by ultra filtration (Pokrovsky et al., 2010; Stolpe et al., 2013). This variation is likely to be driven by changes in the source of the Fe depending on seasonal dynamics in hydrology (Dahlqvist et al., 2007).
4.2 Fe transport capacity and the link to Fe speciation
Nonconservative behavior of Fe was seen in the artificial mixing experiments, as well as in the estuarine gradients sampled, and is consistent with existing literature (Gustafsson et al., 2000; Boyle et al., 1977; Sholkovitz, 1976). Fe transport capacity varied from 0.7 % to 24 % among the rivers. The high Fe transport capacity for most of the rivers studied go along with the existing literature showing that high-latitude DOC-rich rivers exhibit higher Fe-carrying capacities (Krachler et al., 2005; Muller, 2018). DOC was little affected by increasing salinity, as previously observed in high-latitude rivers with high DOC concentrations (Herzog et al., 2017; Forsgren et al., 1996). Nevertheless, the high values of Fe remaining in suspension due to complexation with organic matter at high salinity (0.02–0.50 g L−1) supports the importance of rivers as a source of Fe into the Baltic Sea with an Fe concentration of around 1 µg L−1 (Baltic proper) (Gelting et al., 2010).
Previous studies using size separation and spectrometric methods (Stolpe and Hassellöv, 2007) but also XAS (Herzog et al., 2017) have suggested that mainly Fe (oxy)hydroxide is affected by salinity and selectively lost from suspension by aggregation and sedimentation (Herzog et al., 2017). In contrast, Fe complexed by terrigenous organic matter is supposedly less affected and to a larger extent “surviving” estuarine mixing. Thereby, it can become a source of bioavailable Fe to marine waters (Krachler et al., 2010; Laglera and van den Berg, 2009; Batchelli et al., 2010). The positive correlation between the contribution of Fe-OM (as determined by XAS) and Fe transport capacity (determined in artificial mixing experiments) adds direct support to the idea that organic complexation of Fe enhances the stability across salinity gradients.
In freshwater Fe (oxy)hydroxide is stabilized by surface interactions with organic matter (OM), providing a negative surface charge (Tiller and O'Melia, 1993). With increasing salinity, the surface charge gets neutralized, resulting in reduced colloidal repulsion (Mosley et al., 2003) and formation aggregates containing both Fe and OM. Marine cations, like magnesium and calcium, which neutralize the negatively charged surface groups of the OM, weaken the interaction between colloidal Fe (oxy)hydroxide and OM (Turner and Millward, 2002) and further promote the destabilization of Fe (oxy)hydroxides at increasing salinity. The same cations may favor release and hydrolysis of Fe-OM, by competing for the binding sites of the organic ligands (Fujii et al., 2008). The stability of organically complexed Fe may also be reduced at high ionic strength leading to compression of water and consequent “salting out” of the organic complexes (Turner and Millward, 2002; Turner et al., 2004). Accordingly, Herzog et al. (2017) also identified organically complexed Fe in salinity-induced aggregates especially at high salinities. However, both the selective loss of Fe (oxy)hydroxide at saline conditions and the positive correlation between the relative contribution of Fe-OM and transport capacity in the current study underpin the role of Fe speciation in controlling the fate of Fe across salinity gradients. It would be an advantage to directly measure Fe speciation remaining in suspension in saline samples to see if Fe (oxy)hydroxide is present, but this is currently hindered by methodological limitations. It has been suggested that Fe isotopic ratios may reflect Fe speciation (Ingri et al., 2006; Ilina et al., 2013); however, this remains to be confirmed. Moreover, based on cathodic stripping voltammetry (CSV), ligand concentrations have been found to be in excess of iron concentration, suggesting that organic ligands are complexing the Fe and keeping it in suspension in saline waters (Gledhill and Buck, 2012; Laglera et al., 2011).
Results regarding Fe transport capacity derived from the artificial seawater mixing experiments were in good agreement with the estuarine transects sampled. Theoretically calculated Fe concentrations, based on Fe loss in artificial seawater mixing experiments with river water and the dilution factor, showed only minor deviations from Fe concentrations measured in the Gullmar Fjord. For the Öre estuary, on the other hand, measured Fe concentrations were somewhat higher than the theoretical calculations (Fig. 6). In the low-salinity mixing regime present in the northern Baltic (Bothnian Bay), aggregation may occur without significant sedimentation (Forsgren and Jansson, 1992). This has been observed in the plume of nearby Kalix river and was hypothesized to result from a high organic component of the aggregates, where low specific density may lead to transport of these aggregates far away from the river mouth (Gustafsson et al., 2000). Thus, the centrifugation used to efficiently separate aggregates in the mixing experiments, may overestimate estuarine particle loss in this context. Despite the agreement between measured and theoretically estimated Fe concentrations, the artificial mixing experiments are unlikely to capture all processes that affect the loss of Fe along the natural salinity gradient. In the estuary, photoreduction may affect Fe speciation and affect its fate, as well as the occurrence of ligands produced by marine biota, which may also influence the behavior of riverine Fe. Indeed, the artificial mixing experiments capture the response of riverine Fe to increasing salinity in isolation and how that response depends on Fe speciation.
4.3 Control of spatial variation and flow conditions on Fe speciation
The results of this study showed that Fe speciation is highly variable across spatial scales and during different flow conditions and is further linked to Fe stability across salinity gradients. It is therefore imperative to understand what factors govern Fe speciation. The largest variability in Fe speciation was observed between samples taken along the flow path of Helge river. In the most upstream location, which drains a major peat bog (Svineö), Fe(II) and Fe-OM dominated. This site also showed low oxygen and pH but high DOC concentration – conditions that favor complexation over Fe(III) hydrolysis (Neubauer et al., 2013; Liu and Millero, 2002). The Fe speciation in the stream water close to the Fe sources is thus determined by the properties of the inflowing terrestrial Fe(II)∕Fe(III)-OM complexes and of the conditions when anoxic, hydrated Fe(II) meets oxygenated DOC-rich waters (Sundman et al., 2014). The contribution of Fe(III) and Fe (oxy)hydroxide increased with pH and oxygen saturation along the flow path. This is in agreement with Neubauer et al. (2013), who argued that pH and OM were the main factors controlling Fe speciation in a boreal catchment and explaining the dominance of Fe-OM in wetland-influenced headwaters and increasing Fe (oxy)hydroxide downstream (based on molecular size and chemical equilibrium modeling). The difference in speciation along the flow path may in part be due to organically complexed Fe precipitating as Fe (oxy)hydroxide due to strong hydrolytic tendencies (Karlsson and Persson, 2012) as pH increases and OM declines and in part due to photo reduction of Fe(III)-OM (Fujii et al., 2011; Waite and Morel, 1984; Neubauer et al., 2013). But the difference may also reflect different sources of Fe to the river along the flow path. Thus, while runoff from organic soil layers may bring predominantly Fe-OM to low-order streams (Lydersen et al., 2002; Abesser et al., 2006; Dahlqvist et al., 2007; Sundman et al., 2014), groundwater inflow is more significant further downstream and may bring small Fe (oxy)hydroxides or Fe(II), which is rapidly hydrolyzed in the riparian zone when anoxic groundwater and oxic waters mix (Vasyukova et al., 2010). Hence, the speciation of Fe at the river mouth is determined both by the properties of the inflowing water and the chemical processing along the river flow path.
A consistent pattern was that samples taken in spring, when discharge was higher, showed a larger contribution of Fe-OM than autumn samples from the same river mouths. Temporal variation in river runoff is tightly linked to different hydrogeological pathways (Andersson et al., 2006; Pokrovsky et al., 2006; Neff et al., 2006). During autumn, dominance of groundwater input and longer residence time of ground water should promote input of Fe(II), which rapidly oxidizes to form Fe (oxy)hydroxides in surface water in the absence of high OM concentrations (Dahlqvist et al., 2007). During high discharge, on the other hand, like during spring flood or high precipitation events, organically complexed Fe gets mobilized from the upper soil layers into the river, due to raising water tables and surface runoff (Grabs et al., 2012; Dahlqvist et al., 2007). The lower pH and higher DOC values in our spring samples agree with this reasoning – that during higher discharge flow through organic-rich soil layers has a higher influence on river water chemistry and allows for the formation of more Fe-OM.
It was notable how Fe speciation and Fe stability matched pH across the entire data set. A high contribution of Fe-OM and high stability coincided with low pH across river mouths, along the flow path of the Helge catchment and in the spring samples compared to the autumn samples. pH should exert a strong control on Fe speciation and increasing pH favors precipitation of Fe (oxy)hydroxide due to the strong hydrolytic tendency of Fe(III) (Karlsson and Persson, 2012). Moreover, a low pH may reflect strong influence from organic soils where Fe prevails as Fe-OM on water chemistry, as seen in low-order systems and under high flow conditions (Dahlqvist et al., 2007; Neubauer et al., 2013). On the contrary, a high pH in the river mouth in these boreal systems may reflect a higher influence of groundwater input further downstream, possibly increasing the contribution of Fe (oxy)hydroxide. The increasing pH downstream in parallel with photoreduction and declining DOC concentration may lower the stability by promoting release and hydrolysis of organically complexed Fe originating from organic soils further upstream (Neubauer et al., 2013; Waite and Morel, 1984; Fujii et al., 2011).
The temporal variability within rivers suggests that Fe speciation at a given time is not well predicted by catchment characteristics only. While characteristics such as land cover and soil type most likely affect both quantity and speciation of Fe exported from the catchment, the limited number of rivers and sampling occasions of this study cannot accurately discern such relationships.
The collective results from this study confirmed the existence and wide variability in the contribution of two Fe phases – Fe-OM complexes and Fe (oxy)hydroxides – among the boreal rivers included. It further confirmed that the response of river-borne Fe to increasing salinity differed widely. Interestingly, the differences in stability towards salinity-induced aggregation matched the differences in relative contribution of Fe-OM well across the river mouths, between high and low flow conditions, and along the flow path of a river catchment. Thus, by assessing the Fe speciation by XAS, this study provides direct evidence that Fe-OM enhances survival over estuarine salinity gradients.
This would suggest that high and rising concentrations of Fe from boreal rivers (Kritzberg and Ekström, 2012; Björnerås et al., 2017) may indeed result in increasing export of bioavailable Fe to the Baltic Sea and open waters, where it may limit N-fixation and primary production (Stal et al., 1999; Stolte et al., 2006; Martin and Fitzwater, 1988). Major hydrological events like spring floods and heavy storms have been observed to increase in the Fe concentration by up to a factor of 20 and alter the annual Fe load in northern rivers (Hölemann et al., 2005; Rember and Trefry, 2004; Dahlqvist et al., 2007; Herzog et al., 2019). The hydrogeological conditions during such events (Dahlqvist et al., 2007), may promote a higher contribution of Fe-OM and thus a higher stability during estuarine mixing, resulting an increase in Fe export into the open waters. However, sampling with higher temporal resolution would be required to substantiate such an extrapolation.
Data used to generate all figures are available as tables in the Supplement.
The Supplement contains a high-resolution WT modulus of EXAFS data from the Emån, Alster, and Ljungby river not shown in the paper (Fig. S1); k3-weighted EXAFS spectra and Fourier transformations of all samples and corresponding model fits of all samples (Fig. S3); the relationship between the LCF ratio and CN ratio (Fig. S4); and the relationship between Fe transport capacity at 35 salinity and relative contribution of organically complexed Fe, as assessed by the CN ratio and LCF ratio (Fig. S5). Table S1 shows the k3-weighted Fe K-edge EXAFS fit results for all samples, and Table S2 contains the EXAFS LCF results for the river mouth samples. Tables S3–S5 show the metadata for Figs. 3, 5 and 6. The supplement related to this article is available online at: https://doi.org/10.5194/bg-17-331-2020-supplement.
SDH and ESK conceived of and designed the study. SDH carried out the fieldwork and lab work. SDH, KK and PP performed the XAS analyses and subsequent data treatment. SDH, PP and ESK analyzed the data. SDH wrote the manuscript with support from PP and ESK.
The authors declare that they have no conflict of interest.
Synchrotron work was done at beamline I811, MAX-lab synchrotron radiation source, Lund University, Sweden, and at the high-brilliance X-ray absorption and X-ray emission spectroscopy undulator beamline ID26 of the European Synchrotron Radiation Facility (ESRF, Grenoble). We would like to thank Stefan Carlson for his support on site at the beamline I811. Further, a special thanks to César Nicolás Cuevas for his assistance with the analysis of the XAS data. We thank the Swedish University of Agricultural Sciences (SLU) and Swedish Meteorological and Hydrological Institute (SMHI) for the monitoring data of Swedish rivers. Many thanks to all participants of the COCOA cruise in the Öre estuary in April 2015. A special thanks to Daniel Conley for his efforts in the COCOA project and proofreading of the manuscript. Thanks to Sofia Mebrahtu Wisén for analyzing the samples at the inorganic analytical laboratory.
This research has been supported by the Swedish Research Council (grant no. 2015-05450), the Swedish Research Council Formas (grant no. 207-2010-126), and the European Commission and FORMAS (grant no. 2112932-1).
This paper was edited by Gwenaël Abril and reviewed by two anonymous referees.
Abesser, C., Robinson, R., and Soulsby, C.: Iron and manganese cycling in the storm runoff of a Scottish upland catchment, J. Hydrol., 326, 59–78, 2006.
Andersson, K., Dahlqvist, R., Turner, D., Stolpe, B., Larsson, T., Ingri, J., and Andersson, P.: Colloidal rare earth elements in a boreal river: changing sources and distributions during the spring flood, Geochim. Cosmochim. Ac., 70, 3261–3274, 2006.
Batchelli, S., Muller, F. L., Chang, K.-C., and Lee, C.-L.: Evidence for strong but dynamic iron- humic colloidal associations in humic-rich coastal waters, Environ. Sci. Technol., 44, 8485–8490, 2010.
Björnerås, C., Weyhenmeyer, G. A., Evans, C. D., Gessner, M. O., Grossart, H. P., Kangur, K., Kokorite, I., Kortelainen, P., Laudon, H., and Lehtoranta, J.: Widespread increases in iron concentration in European and North American freshwaters, Global Biogeochem. Cy., 31, 1488–1500, 2017.
Bordas, F. and Bourg, A. C.: A critical evaluation of sample pretreatment for storage of contaminated sediments to be investigated for the potential mobility of their heavy metal load, Water Air Soil Poll., 103, 137–149, 1998.
Boyle, E., Edmond, J., and Sholkovitz, E.: The mechanism of iron removal in estuaries, Geochim. Cosmochim. Ac., 41, 1313–1324, 1977.
Breitbarth, E., Achterberg, E. P., Ardelan, M. V., Baker, A. R., Bucciarelli, E., Chever, F., Croot, P. L., Duggen, S., Gledhill, M., Hassellöv, M., Hassler, C., Hoffmann, L. J., Hunter, K. A., Hutchins, D. A., Ingri, J., Jickells, T., Lohan, M. C., Nielsdóttir, M. C., Sarthou, G., Schoemann, V., Trapp, J. M., Turner, D. R., and Ye, Y.: Iron biogeochemistry across marine systems – progress from the past decade, Biogeosciences, 7, 1075–1097, https://doi.org/10.5194/bg-7-1075-2010, 2010.
Dahlqvist, R., Andersson, K., Ingri, J., Larsson, T., Stolpe, B., and Turner, D.: Temporal variations of colloidal carrier phases and associated trace elements in a boreal river, Geochim. Cosmochim. Ac., 71, 5339–5354, 2007.
Ekström, S. M., Regnell, O., Reader, H. E., Nilsson, P. A., Löfgren, S., and Kritzberg, E. S.: Increasing concentrations of iron in surface waters as a consequence of reducing conditions in the catchment area, J. Geophys. Res.-Biogeo., 121, 479–493, 2016.
Forsgren, G. and Jansson, M.: The turnover of river-transported iron, phosphorus and organic carbon in the Öre estuary, northern Sweden, in: Sediment/Water Interactions, Springer, 585–596, 1992.
Forsgren, G., Jansson, M., and Nilsson, P.: Aggregation and sedimentation of iron, phosphorus and organic carbon in experimental mixtures of freshwater and estuarine water, Estuar. Coast. Shelf S., 43, 259–268, 1996.
Fujii, M., Ito, H., Rose, A. L., Waite, T. D., and Omura, T.: Transformation dynamics and reactivity of dissolved and colloidal iron in coastal waters, Mar. Chem., 110, 165–175, 2008.
Fujii, M., Dang, T., Rose, A. L., Omura, T., and Waite, T.: Effect of light on iron uptake by the freshwater cyanobacterium Microcystis aeruginosa, Environ. Sci. Technol., 45, 1391–1398, 2011.
Funke, H., Scheinost, A., and Chukalina, M.: Wavelet analysis of extended x-ray absorption fine structure data, Phys. Rev. B, 71, 094110, https://doi.org/10.1103/PhysRevB.71.094110, 2005.
Gelting, J., Breitbarth, E., Stolpe, B., Hassellöv, M., and Ingri, J.: Fractionation of iron species and iron isotopes in the Baltic Sea euphotic zone, Biogeosciences, 7, 2489–2508, https://doi.org/10.5194/bg-7-2489-2010, 2010.
Glatzel, P. and Bergmann, U.: High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes – electronic and structural information, Coordin. Chem. Rev., 249, 65–95, 2005.
Gledhill, M. and Buck, K. N.: The organic complexation of iron in the marine environment: a review, Front. Microbiol., 3, 69, https://doi.org/10.3389/fmicb.2012.00069, 2012.
Grabs, T., Bishop, K., Laudon, H., Lyon, S. W., and Seibert, J.: Riparian zone hydrology and soil water total organic carbon (TOC): implications for spatial variability and upscaling of lateral riparian TOC exports, Biogeosciences, 9, 3901–3916, https://doi.org/10.5194/bg-9-3901-2012, 2012.
Gustafsson, Ö., Widerlund, A., Andersson, P. S., Ingri, J., Roos, P., and Ledin, A.: Colloid dynamics and transport of major elements through a boreal river–brackish bay mixing zone, Mar. Chem., 71, 1–21, 2000.
Haese, R. R.: The biogeochemistry of iron, in: Marine Geochemistry, Springer, 241–270, 2006.
Hassellöv, M., Lyvén, B., Haraldsson, C., and Sirinawin, W.: Determination of continuous size and trace element distribution of colloidal material in natural water by on-line coupling of flow field-flow fractionation with ICPMS, Anal. Chem., 71, 3497–3502, 1999.
Herzog, S. D., Persson, P., and Kritzberg, E. S.: Salinity Effects on Iron Speciation in Boreal River Waters, Environ. Sci. Technol., 51, 9747–9755, 2017.
Herzog, S. D., Conrad, S., Ingri, J., Persson, P., and Kritzberg, E. S.: Spring flood induced shifts in Fe speciation and fate at increased salinity, Appl. Geochem., 109, 104385, https://doi.org/10.1016/j.apgeochem.2019.104385, 2019.
Hölemann, J. A., Schirmacher, M., and Prange, A.: Seasonal variability of trace metals in the Lena River and the southeastern Laptev Sea: Impact of the spring freshet, Global Planet. Change, 48, 112–125, 2005.
Hunter, K. A. and Leonard, M. W.: Colloid stability and aggregation in estuaries: 1. Aggregation kinetics of riverine dissolved iron after mixing with seawater, Geochim. Cosmochim. Ac., 52, 1123–1130, 1988.
Ilina, S. M., Poitrasson, F., Lapitskiy, S. A., Alekhin, Y. V., Viers, J., and Pokrovsky, O. S.: Extreme iron isotope fractionation between colloids and particles of boreal and temperate organic-rich waters, Geochim. Cosmochim. Ac., 101, 96–111, https://doi.org/10.1016/j.gca.2012.10.023, 2013.
Ingri, J., Malinovsky, D., Rodushkin, I., Baxter, D. C., Widerlund, A., Andersson, P., Gustafsson, Ö., Forsling, W., and Öhlander, B.: Iron isotope fractionation in river colloidal matter, Earth Planet. Sc. Lett., 245, 792–798, https://doi.org/10.1016/j.epsl.2006.03.031, 2006.
Johnson, K. S., Gordon, R. M., and Coale, K. H.: What controls dissolved iron concentrations in the world ocean?, Mar. Chem., 57, 137–161, 1997.
Karlsson, T. and Persson, P.: Coordination chemistry and hydrolysis of Fe (III) in a peat humic acid studied by X-ray absorption spectroscopy, Geochim. Cosmochim. Ac., 74, 30–40, 2010.
Karlsson, T. and Persson, P.: Complexes with aquatic organic matter suppress hydrolysis and precipitation of Fe(III), Chem. Geol., 322–323, 19–27, https://doi.org/10.1016/j.chemgeo.2012.06.003, 2012.
Karlsson, T., Persson, P., Skyllberg, U., Mörth, C.-M., and Giesler, R.: Characterization of iron (III) in organic soils using extended X-ray absorption fine structure spectroscopy, Environ. Sci. Technol., 42, 5449–5454, 2008.
Kester, D. R., Duedall, I. W., Connors, D. N., and Pytkowicz, R. M.: Preparation of artifical seawater, Limnol. Oceanogr., 12, 176–179, 1967.
Klementev, K.: Statistical evaluations in fitting problems, J. Synchrotron Radiat., 8, 270–272, 2001.
Krachler, R., Jirsa, F., and Ayromlou, S.: Factors influencing the dissolved iron input by river water to the open ocean, Biogeosciences, 2, 311–315, https://doi.org/10.5194/bg-2-311-2005, 2005.
Krachler, R., Krachler, R. F., von der Kammer, F., Suphandag, A., Jirsa, F., Ayromlou, S., Hofmann, T., and Keppler, B. K.: Relevance of peat-draining rivers for the riverine input of dissolved iron into the ocean, Sci. Total Environ., 408, 2402–2408, https://doi.org/10.1016/j.scitotenv.2010.02.018, 2010.
Kraemer, S. M.: Iron oxide dissolution and solubility in the presence of siderophores, Aquat. Sci., 66, 3–18, 2004.
Kritzberg, E. S. and Ekström, S. M.: Increasing iron concentrations in surface waters – a factor behind brownification?, Biogeosciences, 9, 1465–1478, https://doi.org/10.5194/bg-9-1465-2012, 2012.
Kritzberg, E. S., Bedmar Villanueva, A., Jung, M., and Reader, H. E.: Importance of Boreal Rivers in Providing Iron to Marine Waters, PLoS ONE, 9, e107500, https://doi.org/10.1371/journal.pone.0107500, 2014.
Kvashnina, K. O. and Scheinost, A. C.: A Johann-type X-ray emission spectrometer at the Rossendorf beamline, J. Synchrotron Radiat., 23, 836–841, 2016.
Laglera, L. M. and van den Berg, C. M.: Evidence for geochemical control of iron by humic substances in seawater, Limnol. Oceanogr., 54, 610–619, 2009.
Laglera, L. M., Battaglia, G., and van den Berg, C. M.: Effect of humic substances on the iron speciation in natural waters by CLE/CSV, Mar. Chem., 127, 134–143, 2011.
Lalonde, K., Mucci, A., Ouellet, A., and Gélinas, Y.: Preservation of organic matter in sediments promoted by iron, Nature, 483, 198–200, 2012.
Lenstra, W. K., Egger, M., van Helmond, N. A. G. M., Kritzberg, E., Conley, D. J., and Slomp, C. P.: Large variations in iron input to an oligotrophic Baltic Sea estuary: impact on sedimentary phosphorus burial, Biogeosciences, 15, 6979–6996, https://doi.org/10.5194/bg-15-6979-2018, 2018.
Lindström, G., Bartosova, A., Hjerdt, N., and Strömqvist, J.: Uppehållstider i ytvatten i relation tillvattenkvalitetNET, SMHI Rapport Hydrologi No. 199, ett generellt uppskalningsverktyg, 2018.
Liu, X. and Millero, F. J.: The solubility of iron in seawater, Mar. Chem., 77, 43–54, 2002.
Lofts, S., Tipping, E., and Hamilton-Taylor, J.: The chemical speciation of Fe (III) in freshwaters, Aquat. Geochem., 14, 337–358, 2008.
Lydersen, E., Löfgren, S., and Arnesen, R. T.: Metals in Scandinavian surface waters: effects of acidification, liming, and potential reacidification, Crit. Rev. Env. Sci. Tec., 32, 73–295, 2002.
Martin, J. H. and Fitzwater, S. E.: Iron deficiency limits phytoplankton growth in the north-east Pacific subarctic, Nature, 331, 341–343, 1988.
Mosley, L. M., Hunter, K. A., and Ducker, W. A.: Forces between colloid particles in natural waters, Environ. Sci. Technol., 37, 3303–3308, 2003.
Muller, F. L.: Exploring the potential role of terrestrially derived humic substances in the marine biogeochemistry of iron, Front. Earth Sci., 6, 159, https://doi.org/10.3389/feart.2018.00159, 2018.
Neal, C., Lofts, S., Evans, C. D., Reynolds, B., Tipping, E., and Neal, M.: Increasing Iron Concentrations in UK Upland Waters, Aquat. Geochem., 14, 263–288, https://doi.org/10.1007/s10498-008-9036-1, 2008.
Neff, J., Finlay, J., Zimov, S., Davydov, S., Carrasco, J., Schuur, E., and Davydova, A.: Seasonal changes in the age and structure of dissolved organic carbon in Siberian rivers and streams, Geophys. Res. Lett., 33, 2006.
Neubauer, E., Kohler, S. J., von der Kammer, F., Laudon, H., and Hofmann, T.: Effect of pH and stream order on iron and arsenic speciation in boreal catchments, Environ. Sci. Technol., 47, 7120–7128, https://doi.org/10.1021/es401193j, 2013.
Nowostawska, U., Kim, J. P., and Hunter, K. A.: Aggregation of riverine colloidal iron in estuaries: A new kinetic study using stopped-flow mixing, Mar. Chem., 110, 205–210, 2008.
O'day, P. A., Rivera, N., Root, R., and Carroll, S. A.: X-ray absorption spectroscopic study of Fe reference compounds for the analysis of natural sediments, Am. Mineral., 89, 572–585, 2004.
Persson, P. and Axe, K.: Adsorption of oxalate and malonate at the water-goethite interface: molecular surface speciation from IR spectroscopy, Geochim. Cosmochim. Ac., 69, 541–552, 2005.
Pokrovsky, O., Schott, J., and Dupré, B.: Trace element fractionation and transport in boreal rivers and soil porewaters of permafrost-dominated basaltic terrain in Central Siberia, Geochim. Cosmochim. Ac., 70, 3239–3260, 2006.
Pokrovsky, O., Viers, J., Shirokova, L., Shevchenko, V., Filipov, A., and Dupré, B.: Dissolved, suspended, and colloidal fluxes of organic carbon, major and trace elements in the Severnaya Dvina River and its tributary, Chem. Geol., 273, 136–149, 2010.
Raiswell, R., Vu, H. P., Brinza, L., and Benning, L. G.: The determination of labile Fe in ferrihydrite by ascorbic acid extraction: methodology, dissolution kinetics and loss of solubility with age and de-watering, Chem. Geol., 278, 70–79, 2010.
Rember, R. D. and Trefry, J. H.: Increased concentrations of dissolved trace metals and organic carbon during snowmelt in rivers of the alaskan arctic 1, Geochim. Cosmochim. Ac., 68, 477–489, 2004.
Sarkkola, S., Nieminen, M., Koivusalo, H., Laurén, A., Kortelainen, P., Mattsson, T., Palviainen, M., Piirainen, S., Starr, M., and Finér, L.: Iron concentrations are increasing in surface waters from forested headwater catchments in eastern Finland, Sci. Total Environ., 463–464, 683–689, https://doi.org/10.1016/j.scitotenv.2013.06.072, 2013.
Sholkovitz, E.: Flocculation of dissolved organic and inorganic matter during the mixing of river water and seawater, Geochim. Cosmochim. Ac., 40, 831–845, 1976.
Sholkovitz, E., Boyle, E., and Price, N.: The removal of dissolved humic acids and iron during estuarine mixing, Earth Planet. Sc. Lett., 40, 130–136, 1978.
Signorato, R., Solé, V. A., and Gauthier, C.: Performance of the ESRF ID26 beamline reflective optics, J. Synchrotron Radiat., 6, 176–178, https://doi.org/10.1107/S0909049598013971, 1999.
Stal, L. J., Staal, M., and Villbrandt, M.: Nutrient control of cyanobacterial blooms in the Baltic Sea, Aquat. Microb. Ecol., 18, 165–173, https://doi.org/10.3354/ame018165, 1999.
Stolpe, B. and Hassellöv, M.: Changes in size distribution of fresh water nanoscale colloidal matter and associated elements on mixing with seawater, Geochim. Cosmochim. Ac., 71, 3292–3301, https://doi.org/10.1016/j.gca.2007.04.025, 2007.
Stolpe, B., Guo, L., and Shiller, A. M.: Binding and transport of rare earth elements by organic and iron-rich nanocolloids in Alaskan rivers, as revealed by field-flow fractionation and ICP-MS, Geochim. Cosmochim. Ac., 106, 446–462, 2013.
Stolte, W., Balode, M., Carlsson, P., Grzebyk, D., Janson, S., Lips, I., Panosso, R., Ward, C. J., and Graneli, E.: Stimulation of nitrogen-fixing cyanobacteria in a Baltic Sea plankton community by land-derived organic matter or iron addition, Mar. Ecol.-Prog. Ser., 37, 71–82, 2006.
Stumm, W. and Morgan, J.: Aquatic Chemistry, Interscience, NY, 1970.
Sukekava, C., Downes, J., Slagter, H. A., Gerringa, L. J., and Laglera, L. M.: Determination of the contribution of humic substances to iron complexation in seawater by catalytic cathodic stripping voltammetry, Talanta, 189, 359–364, 2018.
Sundman, A., Karlsson, T., and Persson, P.: An experimental protocol for structural characterization of Fe in dilute natural waters, Environ. Sci. Technol., 47, 8557–8564, https://doi.org/10.1021/es304630a, 2013.
Sundman, A., Karlsson, T., Laudon, H., and Persson, P.: XAS study of iron speciation in soils and waters from a boreal catchment, Chem. Geol., 364, 93–102, https://doi.org/10.1016/j.chemgeo.2013.11.023, 2014.
Taylor, S.: Trace element abundances and the chondritic Earth model, Geochim. Cosmochim. Ac., 28, 1989–1998, 1964.
Tiller, C. L. and O'Melia, C. R.: Natural organic matter and colloidal stability: models and measurements, Colloids Surfaces A, 73, 89–102, 1993.
Tipping, E.: The adsorption of aquatic humic substances by iron oxides, Geochim. Cosmochim. Ac., 45, 191–199, 1981.
Turner, A. and Millward, G.: Suspended particles: their role in estuarine biogeochemical cycles, Estuar. Coast. Shelf S., 55, 857–883, 2002.
Turner, A., Millward, G. E., and Le Roux, S. M.: Significance of oxides and particulate organic matter in controlling trace metal partitioning in a contaminated estuary, Mar. Chem., 88, 179–192, 2004.
van Hulten, M., Middag, R., Dutay, J.-C., de Baar, H., Roy-Barman, M., Gehlen, M., Tagliabue, A., and Sterl, A.: Manganese in the west Atlantic Ocean in the context of the first global ocean circulation model of manganese, Biogeosciences, 14, 1123–1152, https://doi.org/10.5194/bg-14-1123-2017, 2017.
Vasyukova, E., Pokrovsky, O., Viers, J., Oliva, P., Dupré, B., Martin, F., and Candaudap, F.: Trace elements in organic-and iron-rich surficial fluids of the boreal zone: Assessing colloidal forms via dialysis and ultrafiltration, Geochim. Cosmochim. Ac., 74, 449–468, 2010.
Vilgé-Ritter, A., Rose, J., Masion, A., Bottero, J.-Y., and Lainé, J.-M.: Chemistry and structure of aggregates formed with Fe-salts and natural organic matter, Colloid. Surface. A, 147, 297–308, 1999.
Waite, T. D. and Morel, F. M.: Photoreductive dissolution of colloidal iron oxides in natural waters, Environ. Sci. Technol., 18, 860–868, 1984.
Wällstedt, T., Björkvald, L., and Gustafsson, J. P.: Increasing concentrations of arsenic and vanadium in (southern) Swedish streams, Appl. Geochem., 25, 1162–1175, 2010.
Waychunas, G. A., Kim, C. S., and Banfield, J. F.: Nanoparticulate iron oxide minerals in soils and sediments: unique properties and contaminant scavenging mechanisms, J. Nanopart. Res., 7, 409–433, 2005.
Webb, S.: SIXpack: a graphical user interface for XAS analysis using IFEFFIT, Phys. Scripta, T115, 1011–1014, 2005.
Weyhenmeyer, G. A., Prairie, Y. T., and Tranvik, L. J.: Browning of Boreal Freshwaters Coupled to Carbon-Iron Interactions along the Aquatic Continuum, PLoS ONE, 9, e88104, https://doi.org/10.1371/journal.pone.0088104, 2014.
Yu, C., Virtasalo, J. J., Karlsson, T., Peltola, P., Österholm, P., Burton, E. D., Arppe, L., Hogmalm, J. K., Ojala, A. E., and Åström, M. E.: Iron behavior in a northern estuary: Large pools of non-sulfidized Fe (II) associated with organic matter, Chem. Geol., 413, 73–85, 2015.
Zabinsky, S., Rehr, J., Ankudinov, A., Albers, R., and Eller, M.: Multiple-scattering calculations of X-ray-absorption spectra, Phys. Rev. B, 52, 2995, https://doi.org/10.1103/PhysRevB.52.2995, 1995.