the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 10 Feb 2020
| 10 Feb 2020
High-pH and anoxic conditions during soil organic matter extraction increases its electron-exchange capacity and ability to stimulate microbial Fe(III) reduction by electron shuttling
Yuge Bai
Edisson Subdiaga
Stefan B. Haderlein
Heike Knicker
Andreas Kappler
Soil organic matter (SOM) is redox-active, can be microbially reduced, and transfers electrons in an abiotic reaction to Fe(III) minerals, thus serving as an electron shuttle. The standard procedure to isolate organic matter (OM) from soil involves the use of alkaline and acidic solutions and the separation of humic acids (HAs) and fulvic acids (FAs). This process potentially leads to unwanted changes in SOM chemical and redox properties. To determine the effects of extraction conditions on the redox and electron-shuttling properties of SOM extracts, we prepared HA, FA, and water-extractable organic matter (OM) extracts, applying either a combination of 0.1 M NaOH and 6 M HCl or ultrapure water (pH 7), from soil samples collected from the subsoil (0–15 cm, A horizon, pH 6.5–6.8) in Schönbuch forest, Baden-Württemberg, Germany. Both chemical extractions (NaOH∕HCl) and water extractions were done in separate experiments under either oxic or anoxic conditions. Furthermore, we applied the NaOH∕HCl treatment to a subsample of the water-extractable OM to separate HA and FA from the water-extractable OM. When comparing the amount of carbon extracted from soil by different extraction methods, we found that FA and HA chemically extracted from the soil can make up to 34 %–40 % of the soil organic carbon pool while the water-extractable OM only represents 0.41 %–2.74 % of the total soil organic carbon. The higher extraction efficiency of the chemical extraction is probably due to the deprotonation of carboxyl and phenol functional groups under high pH. Anoxic extraction conditions also led to more extracted carbon. For water-extractable OM, 7 times more C was extracted under anoxic conditions compared to oxic conditions. This difference was probably due to the occurrence of microbial reduction and dissolution of Fe(III) minerals in the soil during the anoxic water extraction and thus the concomitant release of Fe(III) mineral-bound organic matter. To compare the redox activity of different SOM extracts, the electron-exchange capacity (EEC) of all extracted HA, FA, and water-extractable OM was analyzed and our results showed that, under anoxic extraction conditions, the HA chemically isolated from the water-extractable OM had 2 times higher EEC values compare to the water-extractable OM itself, suggesting the potential formation of redox-active aromatic functional groups during the extraction with NaOH under anoxic conditions by condensation reactions between amino acids, aldehydes, and hydroxyl- and catechol-containing molecules. We also performed a microbial Fe(III) reduction experiment with all extracts and found that higher EEC of extracts in turn resulted in a higher stimulation of microbial Fe(III) mineral reduction by electron shuttling, i.e., faster initial Fe(III) reduction rates, and in most cases also in higher reduction extents. Our findings suggest that OM extracted with water at neutral pH should be used to better reflect environmental SOM redox processes in lab experiments and that potential artefacts of the chemical extraction method and anoxic extraction condition need to be considered when evaluating and comparing abiotic and microbial SOM redox processes.
- Article
(2408 KB) - Full-text XML
-
Supplement
(2939 KB) - BibTeX
- EndNote





Soil organic matter (SOM) contains more organic carbon than the sum of the atmosphere and living plants (Fischlin et al., 2007) and can influence greenhouse gas emission, plant growth, and water quality (Lal, 2004; Marin-Spiotta et al., 2014). Studying SOM is challenging because it needs to be separated from other soil components before doing laboratory experiments (Lehmann and Kleber, 2015). One of the most commonly used methods is a chemical extraction of humic substances (HSs) at pH > 12 (Achard, 1786). Although the concept of HS as high-molecular-weight molecules formed by degradation and repolymerization of biomolecules has been challenged by seeing SOM as a continuum of progressively decomposing organic compounds (Lehmann and Kleber, 2015), HS extraction is still applied by many laboratories and the extracted HSs are still widely used as a proxy for SOM. Briefly, HSs are extracted by adjusting the pH to >12 using NaOH, followed by acidification of the alkaline extract to pH < 2 to separate humic acids (HAs) from fulvic acids (FAs) (Achard, 1786). Ion exchange resins, dialysis, and even hydrofluoric acid (HF) treatment are used to further purify the extracts (IHSS, 2019). Concerns regarding the effectiveness of this harsh chemical extraction method were already raised in 1888 (van Bemmelen, 1888) and have lasted until today (Lehmann and Kleber, 2015; Kleber and Lehmann, 2019).
It has been shown that alkaline extraction influences the chemical composition and the content of redox-active quinoid moieties of the extracted SOM (Piccolo, 1988; Engebretson and Von Wandruszka, 1999). Participation in redox reactions is a key property of SOM and relevant for many biogeochemical processes in the environment (Murphy, 2014). For example, under anoxic conditions, SOM can accept electrons from microorganisms, transfer electrons to other electron acceptors such as Fe(III) minerals, and be reoxidized to accept electrons again from microorganisms (Lovley et al., 1996; Kappler et al., 2004; Bauer and Kappler, 2009; Wolf et al., 2009). This electron-shuttling process, which is facilitated by SOM, can significantly increase microbial reduction rates of poorly soluble Fe(III) minerals (Lovley et al., 1996; Jiang and Kappler, 2008), enable microbial reduction of otherwise inaccessible Fe(III) minerals (Lovley et al., 1998) and stimulate indirect reduction of minerals that are spatially separated from the bacteria (Lies et al., 2005). Highly purified FA and HA are used in most electron-shuttling studies to represent SOM (Lovley et al., 1998; Lovley and Blunt-Harris, 1999; Lies et al., 2005; Bauer and Kappler, 2009; Wolf et al., 2009; Klupfel et al., 2014). However, currently it is not known if and to which extent the SOM electron-shuttling capacity is based on protocol-induced changes caused by the harsh chemical isolation procedure. Therefore, studies that compare the stimulating effects of SOM extracted with either the traditional chemical extraction method or with water at neutral pH conditions on the microbial Fe(III) reduction are needed.
Piepenbrock and co-authors extracted SOM from a forest soil at circumneutral pH using water (Piepenbrock et al., 2014). Compared to chemically extracted Pahokee Peat humic acids (PPHAs), the extracted SOM had a significantly lower reducing capacity (µeq g C−1), which was calculated from the concentration of reduced Fe(II) after the abiotic reaction of PPHA and SOM with Fe(III) citrate. This potentially indicates different types and proportions of functional groups in these samples. However, due to the different origin of the extracted soils, it remains unclear whether and to which extent the differences in reducing capacities of the SOM extract and PPHA was caused by the chemical extraction methods. Furthermore, in this study the water extraction was conducted only under oxic conditions. Although it is known that the presence of O2 causes oxidation of certain organic compounds under alkaline conditions and therefore chemical extraction with NaOH should be conducted under anoxic conditions (Bauer and Kappler, 2009; Maurer et al., 2010), it remains unclear whether and how the presence of O2 influences the abundance of different (redox-active) functional groups and therefore the redox activity of the water-extracted organic compounds under neutral pH.
To determine the effect of these chemicals on the SOM redox properties, we extracted OM from a forest soil using several methods (Fig. 1). The first was the traditional chemical extraction method (1 M NaOH followed by 6 M HCl) yielding HA and FA under either oxic or anoxic conditions. The second was OM extraction by ultrapure water at neutral pH (water-extractable OM) under either oxic or anoxic conditions. Additionally, we treated the water-extractable OM with NaOH and HCl to further separate HA and FA from the water-extractable OM (also under either oxic or anoxic conditions). We analyzed the electron-accepting capacity (EAC, i.e., the number of electrons that can be accepted), the electron-donating capacity (EDC, i.e., the number of electrons that can be donated by the OM), and the electron-exchange capacity (the sum of EAC and EDC) of all extracted water-extractable OM, FA, and HA fractions. To further compare their electron-shuttling capacity, we performed a microbial Fe(III) mineral reduction experiment with all of the different extracts. The goals of this study were, first, to identify the effects of alkali and oxygen on the EEC values of the water-extractable OM, FA, and HA samples and, second, to compare the rates and extents of microbial ferrihydrite reduction in the presence of the different extracts.
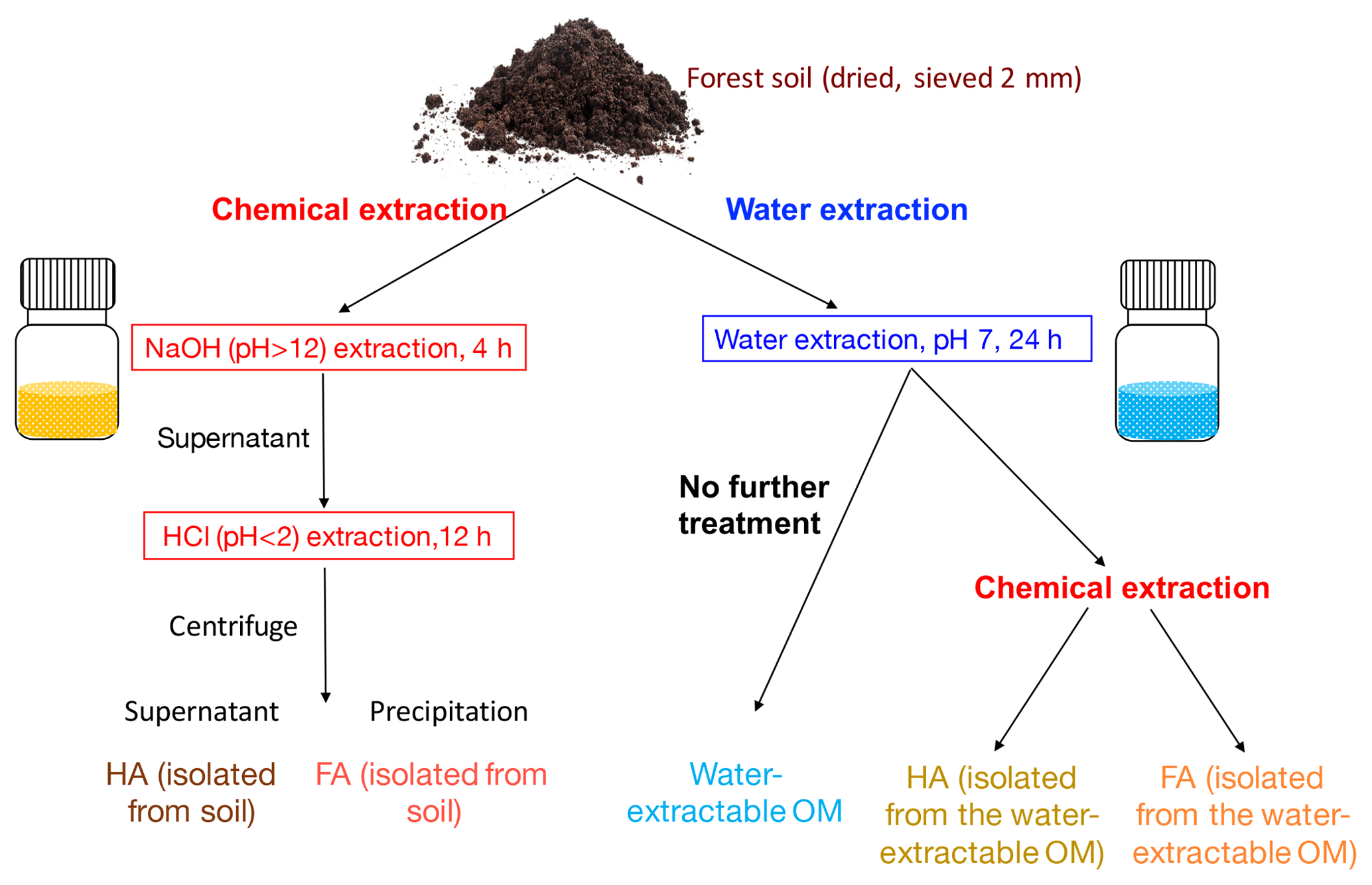
Figure 1Schematic diagram of the soil organic matter (SOM) extraction process. Forest soil samples (Schönbuch forest, Baden-Württemberg, Germany) were dried under 30 ∘C and ground to pass through a 2 mm sieve. To isolate FA and HA from soil, 100 g of soil was incubated with 400 mL 0.1 M NaOH (pH 12) for 4 h; after centrifugation, the supernatant was acidified by HCl to pH < 2. HA was then precipitated out to be separated from FA, and we define these two extracts as HA (isolated from soil) and FA (isolated from soil). For water extraction, 100 g of soil was incubated with 400 mL ultrapure water (<18.2 MΩ cm; Milli-Q, Millipore) at a pH of 7. pH was monitored during the extraction and it remained stable (range between 7.02 and 7.33). The slurry was centrifuged after 24 h; we define the supernatant as water-extractable OM. A 200 mL aliquot of the supernatant containing the water-extractable OM was amended with 1 M NaOH until a pH of 12. After 4 h of incubation, the pH of the solution was adjusted to <2 by 6 M HCl to precipitate HA and to separate FA, and these two fractions are HA (isolated from water-extractable OM) and HA (isolated from water-extractable OM). All of the extractions were conducted under both oxic and anoxic conditions.
2.1 Soil organic matter (SOM) extraction
Top soil (0–15 cm) without leaf litter from the A horizon was collected from the Schönbuch forest, Baden-Württemberg, Germany. The forest is dominated by beech with populations of oak, spruce, and bald cypress and the soil is qualified as vertic cambisol (World Reference Base for Soil, WRB, Schad et al., 2014). Soil was dried (30 ∘C), ground to pass through a 2 mm sieve by an automatic soil grinder (ball mill, Fritsch, Germany) and stored in the dark at 4 ∘C. Chemical extraction of FA and HA was modified from the IHSS protocol (IHSS, 2019) as follows. In the IHSS protocol, soil samples are incubated with 0.1 M HCl with a ratio of 10 mL of liquid per gram of dry soil, and after 1 h of extraction and XAD-8 resin separation, a fraction called “FA_extract_1” is collected. In our experiment, we did not extract this fraction to avoid using XAD-8 separation. As shown in Fig. 1, 100 g of soil was incubated with 400 mL of 0.1 M NaOH (pH 12) for 4 h (overhead shaker, 90 rpm, 25 ∘C). The slurry was centrifuged (3528×g, 30 min) and the supernatant was acidified (pH 2) by 6 M HCl to separate FA (dissolved in the supernatant) and HA (precipitated). Within the present study we define these extracts as FA (isolated from soil) and HA (isolated from soil). Water-extractable OM was prepared following Piepenbrock et al. (2014) (Fig. 1). 100 g of soil was incubated with 400 mL of ultrapure water (resistivity = 18.2 MΩ cm, 25 ∘C; Milli-Q, Millipore) at a pH of 7 (pH was monitored during the extraction and it ranged between 7.02 and 7.33). The pH of the water extraction was adjusted to 7 to avoid any possible artefacts resulting from further pH adjustment prior to analyses and experiments that require the sample pH to be 7 such as the electrochemical analysis or the microbial Fe(III) reduction. The slurry was centrifuged after 24 h; we collected and defined the supernatant as water-extractable OM. A 200 mL aliquot of the supernatant containing the water-extractable OM was amended with 0.1 M NaOH until a pH of 12. After 4 h of incubation, the pH of the solution was adjusted to <2 by 6 M HCl to precipitate HA and to separate FA; this FA and HA are defined as FA (isolated from water-extractable OM) and HA (isolated from water-extractable OM). Anoxic chemical extractions (NaOH∕HCl) and water extractions were performed in a N2-filled anoxic glove box; filtration (0.45 µm, polyethersulfone membrane (PES), Millipore, Germany) was used instead of centrifugation to remove remaining soil after the extraction. All collected extracts were freeze-dried under oxic conditions and stored (4 ∘C, dark) until use.
2.2 Organic carbon quantification
Dissolved organic carbon (DOC) of water-extractable OM and FA isolated from soil was quantified using a DOC analyzer (model 2100S, Analytik Jena, Germany) directly from the extracted solutions after passing through a 0.45 µm syringe filter (polyethersulfone membrane (PES), Millipore, Germany). The total organic carbon (TOC) of all extracted HAs and the FA isolated from the water-extractable OM were quantified using a TOC analyzer (model 2100S, Analytik Jena, Germany) from the freeze-dried powders.
2.3 Nuclear magnetic resonance (NMR) measurements
NMR analysis was conducted from freeze-dried water-extractable OM, HA, and FA samples. Solid-state 13C NMR spectra were obtained with a Bruker AVANCE III HD 400 MHz wide board operating at a frequency of 100.63 MHz using zirconium rotors of 4 mm OD with Kel-F caps. The cross-polarization magic-angle spinning (CPMAS) technique was applied during magic-angle spinning of the rotor at 14 kHz. A ramped 1H pulse was used in order to circumvent spin modulation of Hartmann–Hahn conditions. A contact time of 1 ms and a 90∘ 1H-pulse width of 2.2 µs were used for all spectra. The 13C-chemical shifts were calibrated to tetramethylsilane (0 ppm) and were calibrated with glycine (176.04 ppm). The aromaticity of samples was calculated following a previous study (Abelmann et al., 2005).
2.4 Specific UV absorbance at 254 (SUVA254) analysis
SUVA254 analysis was conducted from water-extractable OM, HA, and FA solutions dissolved in Milli-Q water at concentrations of 10 mg C L−1. All solutions were filtered with a 0.45 µm syringe filter (polyethersulfone membrane (PES), Millipore, Germany), and the dissolved organic carbon (DOC) concentration of all samples was analyzed prior to the SUVA254 analysis (DOC analyzer, model 2100S, Analytik Jena, Germany). The SUVA254 values of all samples were measured in a 1 cm rectangular quartz cuvette with a fluorescence spectrophotometer (FluoroMax-4, Jobin Yvon–SPEX instruments, New Jersey, USA). The final SUVA254 values of all extracts were calculated with Eq. (1):
where UV254 is the absorbance at 254 nm and there is a 1 cm optical path length.
2.5 Microwave plasma atomic emission spectrometer (MP-AES) analysis
Metal contents were analyzed by a microwave plasma atomic emission spectrometer (MP-AES) (4100, Agilent Inc., Santa Clara, CA, USA) in the extracted water-extractable OM, HA, and FA samples. Prior to the MP-AES analysis, a 0.5 g sample was digested with 10 mL 2 % HNO3 in a microwave oven at 190 ∘C (800 W) for 10 min; after cooling down to room temperature, it was centrifuged for 10 min at 28 649 ×g and the supernatant was used for the analysis. The data for FA (isolated from soil, oxic) and HA (isolated from water-extractable OM, oxic) are missing due to the lack of enough samples. The unit of all metal concentrations is milligrams per kilogram, blank means the concentration of the corresponding metal is too low to be detected.
2.6 Electrochemical analysis
Electrochemical analysis followed the method described by Aeschbacher et al. (2010). Freeze-dried extracts (powders) were dissolved in 100 mM of phosphate buffer (pH 7) at a concentration of 100 mg C L−1. After overnight agitation at 300 rpm at room temperature, samples were filtered through 0.22 µm syringe filters (mixed cellulose ester (MCE), Millipore, Germany). All preparations and measurements were conducted in an anoxic glove box. The number of electrons transferred to and from all extracts was quantified by integration of reductive and oxidative current responses after baseline correction in mediated electrochemical reduction (MER; at V) and mediated electrochemical oxidation (MEO; V) with 1′1-ethylene-2,2′-bipyridyldiylium di-bromide (DQ) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) as electron transfer mediators, respectively. To obtain the EAC and EDC values, the integrated current response was normalized to the measured DOC of all extracts prior to the EEC analysis (DOC (mg C L−1), DOC analyzer, model 2100S, Analytik Jena, Germany) as shown in Eqs. (2) and (3):
where Ired and Iox ([A]) are baseline-corrected reductive and oxidative currents in MER and MOR, respectively (F = Faraday constant).
2.7 Microbial Fe(III) reduction experiment and calculation of microbial Fe(III) reduction rates
Solutions of organic matter extracts for the microbial Fe(III) reduction experiment were prepared by dissolving freeze-dried powders in 50 mM phosphate buffer (pH 7.0–7.2) at 500 mg C L−1, agitation overnight (300 rpm, room temperature), and filtration and sterilization (0.22 µm syringe filters, mixed cellulose ester (MCE), Millipore, Germany), as described before (Jiang and Kappler, 2008). Although the chosen phosphate concentration is higher than typically observed in nature and can potentially lead to the formation of Fe(II) phosphate minerals (e.g., vivianite) during our microbial Fe(III) reduction experiment, this phosphate buffer was chosen to enable comparison of our study to previous studies (Jiang and Kappler, 2008; Bauer and Kappler, 2009; Klupfel et al., 2014; Piepenbrock et al., 2014). All solutions were deoxygenated three times (each time with 3 min vacuum and 3 min N2 flushing) and stored in dark bottles to avoid photochemical reactions. Ferrihydrite was prepared as described before (Amstaetter et al., 2012) and stored no more than 2 months (4 ∘C) before use.
Shewanella oneidensis MR-1 cells from a frozen stock were streaked on oxic lysogeny broth (LB) agar plates (10 g L−1 peptone, 5 g L−1 yeast extract, 10 g L−1 NaCl, and 15 g L−1 agar). Colonies were transferred to liquid LB medium and incubated at 30 ∘C for 14 h, harvested by centrifugation (10 min, 8602×g) and then washed three times with anoxic Shewanella Basal Medium (SBM) medium (0.225 g L−1 K2HPO4, 0.225 g L−1 KH2PO4, 0.46 g L−1 NaCl, 0.225 g L−1 (NH4)2SO4, 2.18 g L−1 Na-lactate, 0.117 g L−1 MgSO4•7H2O, 2.38 g L−1 HEPES, pH 7.2–7.5). For the Fe(III) reduction experiments, washed cells were added at a final concentration of 107 cells mL−1 to solutions of water-extractable OM, FA, HA (50 mg C L−1), and ferrihydrite (15 mM Fe(III)) in SBM medium (phosphate in the SBM medium was 5 mmol L−1). AQDS, i.e., 2,6-anthraquinone disulfonate, a quinone model compound commonly used in electron-shuttling studies that can significantly increase the rates of microbial Fe(III) reduction, was used as 100 µmol L−1 in our experiments as a reference for a significant stimulation of Fe(III) reduction by our extracted OM via electron shuttling. The headspace was flushed with N2 and the bottles were incubated in the dark (30 ∘C). At each sampling point, an 100 µL aliquot was taken from each bottle, acidified and incubated with 900 µL of 1 M HCl for 1 h to facilitate mineral dissolution, and centrifuged (28 649 × g, 5 min), and the total Fe(II) concentration was quantified with the spectrophotometric ferrozine assay in a microtiter plate (Stookey, 1970; Hegler et al., 2008). The fastest reduction rates of the microbial Fe(III) reduction experiments were calculated as shown and explained in the Supplement (Fig. S3).
3.1 Quantity of soil organic carbon extracted by different methods and characterization of extracted OM, HA, and FA
Comparison of different extraction methods revealed that the amount of soil-extracted carbon varied depending on the presence or absence of O2 during the extraction and on the type of extraction liquid (Table 1). Extraction with H2O at neutral pH under oxic conditions followed by NaOH and HCl treatment to separate HA and FA in the water-extractable OM yielded 0.036, 0.021, and 0.014 g C in the water-extractable OM, FA, and HA fractions, respectively, corresponding to 0.41 %, 0.24 %, and 0.15 % of the total carbon present in the soil. In contrast, anoxic water extraction significantly increased the fraction of extracted carbon to 0.234, 0.146, and 0.079 g C in the water-extractable OM, FA, and HA fractions, respectively, corresponding to 2.74 %, 1.69 %, and 0.90 % of the total soil C. Chemical extraction directly from soil using NaOH and HCl under oxic conditions yielded 1.451 g in the extracted FA (17.0 % of the total carbon present in the soil) and 1.450 g C in HA (17.0 % of the total carbon present in the soil). Conducting the same chemical extraction from soil under anoxic conditions led to a higher percentage of extracted carbon for FA (20.7 %) and HA (22 %) than under oxic conditions.
Table 1Total dissolved organic carbon (DOC) concentration of the water-extractable OM and FA isolated from soil, total organic carbon in all extracted OM, HA, and FA fractions, and the calculated percentage of carbon extracted from the soil. DOC of water-extractable OM and FAs isolated from soil fractions was quantified directly from their extracted solutions, whereas the FA isolated from water-extractable OM and all of the HA extracts were first freeze-dried and then redissolved in Milli-Q water at a pH of 7 for analysis. The difference between total carbon (mg) of water-extractable SOM and the sum of total carbon (mg) of FA and HA isolated from the water-extractable OM (under oxic and anoxic conditions) is due to the loss of material during the isolation of FA and HA. Values are means ± standard deviation (SD) of triplicates. An unpaired two-sided T test was done to analyze the statistical significance of each pair of samples that was treated with the same extraction method but under different conditions (i.e., FA isolated from water-extractable OM, oxic, was compared to FA isolated from water-extractable OM, anoxic)

a TOC of all HA extracts and of the FA isolated from the water-extractable OM was directly quantified from the freeze-dried powders. The carbon content of all other liquid samples was determined as DOC, measured directly from the solutions after passing through 0.45 µm syringe filter. b Total organic carbon content in the extract was directly quantified from the freeze-dried samples of FA isolated from the water-extractable OM (oxic, anoxic) and of the HA extracts. For the other extract solutions, the total organic carbon was calculated by DOC (mg C L−1) times the volume of the extracted solution (L). c Percentage of carbon extracted from soil is equal to the total carbon extracted (mg g−1) divided by soil carbon content (8.54 mg g−1). d Specific UV absorbance 254 nm (SUVA254) = UV254 × DOC (mg C L−1); b is the optical path length in centimeters (1 cm in this experiment). * The percentage of carbon extracted from soil of the samples is significantly different (n=2, two-sided t test, P<0.05).
Specific UV absorbance at 254 nm (SUVA254) indicates the aromaticity of the extracted OM, FA, and HA (Table 1). Water-extractable OM showed a SUVA254 value of 0.018 mg−1 C cm−1 under oxic conditions, and the value increased to 0.027 mg−1 C cm−1 when extracted under anoxic conditions. Similarly, for both FA isolated from water-extractable OM and FA isolated from soil, the SUVA254 values were higher under anoxic conditions than under oxic conditions. HA extracts isolated from the water-extractable OM under oxic conditions showed a SUVA254 value of 0.068 mg−1 C cm−1, higher than the 0.018 mg−1 C cm−1 of the water-extractable OM itself obtained under oxic conditions. A higher SUVA254 value for the HA isolated from the water-extractable OM (0.207 mg−1 C cm−1) than for the water-extractable OM itself (0.027 mg−1 C cm−1) was also found under anoxic extraction conditions. A two-way ANOVA statistical analysis revealed that both oxic versus anoxic conditions and the extraction method (neutral pH water versus chemical extraction) resulted in different SUVA254 values at a significance level of P<0.05 (Table S1). In general, anoxic conditions and the chemical extraction method led to higher SUVA254 values of the extracts, suggesting that these extracts had a higher degree of aromaticity (Korshin et al., 1997). 13C-NMR analysis of extracted OM, FA, and HA (Fig. 2) confirmed higher contents of aromatic carbon in samples subject to chemical extraction or anoxic conditions.
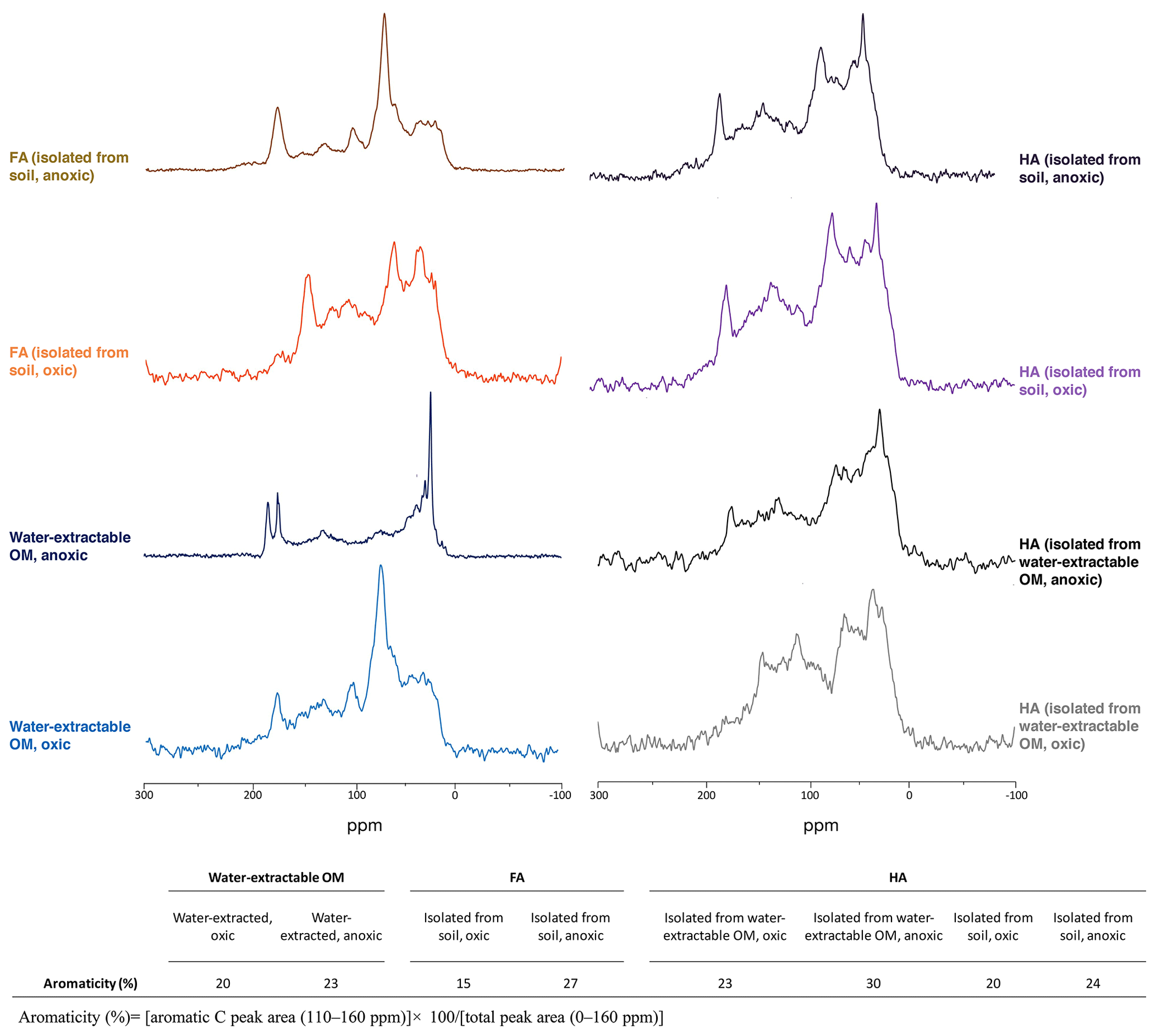
Figure 213C-NMR spectra of water-extractable OM, FA, and HA. All spectra were collected from freeze-dried extracts and the aromaticity of samples was calculated as the percentage of aromatic C peak area to the total peak area. The relative intensity distributions of specific chemical shifts are shown in the Supplement, Table S2. Spectra for FA isolated from water-extractable OM cannot be presented due to the lack of an adequate amount of sample for analysis.
Furthermore, after 24 h water extraction of OM, we found a maximum Fe(II) concentration of 3 mmol L−1 in the water-extractable OM solution. Although, as shown in Table S3, more than 90 % of the Fe was removed by filtration (0.22 µm, mixed cellulose ester (MCE), Millipore, Germany) and around 30 % of the remaining Fe(II) was oxidized to Fe(III) during the oxic freeze-drying process, there was still 15–123 µmol L−1 total Fe present in the water-extractable OM and FA or HA isolated from it. This Fe is potentially redox-active and can contribute to the redox properties of the extracted OM. For example, a previous study showed that 4 mg L−1 of Fe(III) yielded an absorbance value of 0.65 cm−1 at the 254 nm wavelength (Weishaar et al., 2003). Therefore, we believe that the high SUVA254 value of HA isolated from the water-extractable OM compared to SUVA254 values of HA shown in previous studies could be caused by the presence of Fe(II) and Fe(III) in the sample due to the microbial Fe(III) reduction that occurred under the anoxic extraction conditions. However, since this Fe is an integral part of the OM in the environment and we were interested in determining the role of environmentally relevant OM extracts in electron shuttling, we decided not to further purify the extracts (also because this probably would have changed the properties of the present redox-active organic matter).
3.2 Redox properties of extracted water-extractable OM, FA, and HA
We used mediated electrochemical reduction and oxidation to quantify the EAC, EDC, and thus EEC of all water-extractable OM, FA, and HA extracts (Fig. 3). Based on the Fe content we calculated the contribution of the Fe to the redox properties of all extracts (Table S3). A two-way ANOVA statistical analysis revealed that both the extraction condition (anoxic versus oxic) and the extraction method (neutral pH water versus NaOH) resulted in significantly different EEC values (P<0.05; Table S4). The EEC of water-extractable OM obtained under oxic conditions was 32 µmol e− mmol C−1 (with ca. 4 µmol e− mmol C−1 from Fe), whereas when extracted anoxically, it increased to 44 µmol e− mmol C−1 (with 14.8 µmol e− mmol C−1 from Fe). Higher EEC values under anoxic compared to oxic extraction conditions were also observed for all extracted FA: for FA isolated oxically from the water-extractable OM, the EEC was 13 µmol e− mmol C−1 (2.3 µmol e− mmol C−1 from Fe), while it increased to 24 µmol e− mmol C−1 (2.7 µmol e− mmol C−1 from Fe) when FA was isolated anoxically from the water-extractable OM. The EEC of FA isolated from soil under anoxic conditions was 33 µmol e− mmol C−1 higher than FA isolated from soil under oxic conditions. Similar to FA, for the HA isolated from water-extractable OM, the EEC values increased from 15 µmol e− mmol C−1 (1.9 µmol e− mmol C−1 from Fe) under oxic conditions to 83 µmol e− mmol C−1 (7.3 µmol e− mmol C−1 from Fe) under anoxic conditions. For HA isolated from soil, EEC values increased from 40 µmol e− mmol C−1 under oxic conditions to 127 µmol e− mmol C−1 under anoxic conditions.
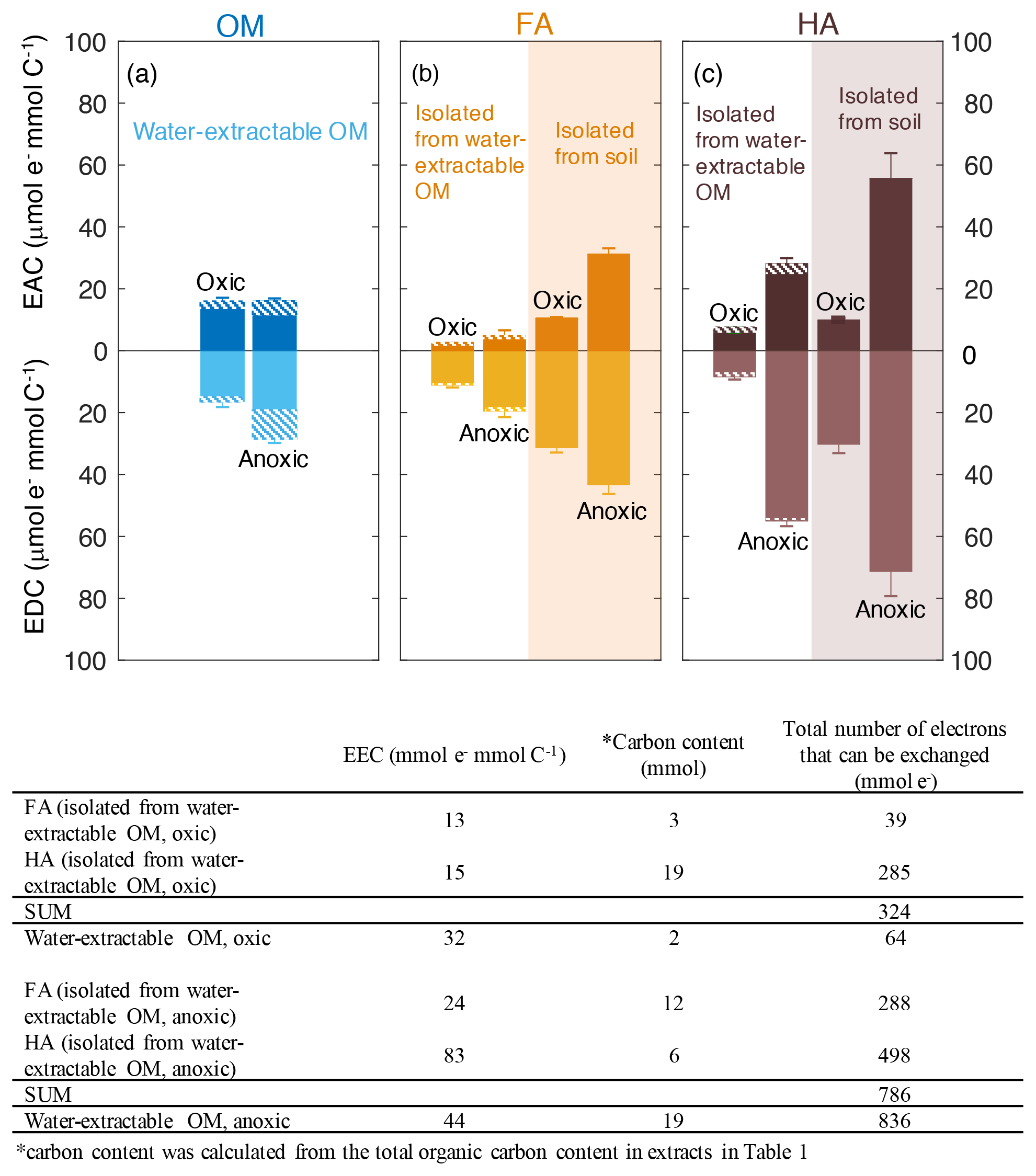
Figure 3Electron-exchange capacity (EEC), the sum of EAC (electron-accepting capacity) and EDC (electron-donating capacity) of water-extractable OM (a), FA (b), and HA (c) extracted from Schönbuch forest soil under oxic or anoxic conditions. Areas with white background represent FA and HA isolated from water-extractable OM, whereas orange and purple shaded areas represent FA and HA isolated from soil, respectively. Hashed areas represent the EAC or EDC contribution stemming from redox-active Fe(III) and Fe(II) ions in the samples. The integrated current response was normalized to the DOC of all extracts. Despite the contribution of Fe to the redox activity, we decided to normalize the EAC and EDC values to C content because in most cases the Fe contribution was small and because normalizing the EAC and EDC values to total weight of material would be misleading since the fraction of inorganic, non-redox active constituents (e.g., Mg2+, Ca2+ ions from salts present in the soil) varies significantly between the different extracts (Table S5). Error bars indicate the standard deviations of at least four replicates. Two-way ANOVA statistical analysis was conducted and the result suggested the measured EEC values of the extracts were significantly different from each other (P<0.005, Table S4). The table underneath the figure shows the recovery of total number of electrons that can be exchanged (that means transferred from Fe(III)-reducing bacteria to the OM, or from the OM to Fe(III) minerals) by water-extractable OM before and after the chemical isolation of HA and FA.
The total number of electrons that can be exchanged (that means transferred from Fe(III)-reducing bacteria to the OM, or from the OM to Fe(III) minerals) by water-extractable OM before and after the chemical separation of FA and HA from this water-extractable OM was also calculated (the recovery of EEC) under both oxic and anoxic conditions (Fig. 2). For the extracts obtained under anoxic conditions, the sum of total exchangeable electron values of the FA and HA isolated from water-extractable OM (786 µmol e−) was almost identical to that of water-extractable OM itself (836 µmol e−). In contrast, under oxic conditions, the sum of the EEC values of the FA and HA separated from the water-extractable OM was 324 µmol e−, ca. 5 times higher than the EEC value of the water-extractable OM (64 µmol e−). This confirms that the traditional chemical extraction procedure conducted under oxic conditions strongly enhances the redox capacity of the samples.
In addition to the EEC that represents the total amount of electrons that can be stored by the extracted organic compounds, the individual EAC and EDC values can be used to characterize the redox state of the water-extractable OM, HA, and FA. The EDC and EAC quantify how many electrons are already stored in the molecules (EDC) and how many electrons can still be taken up by functional groups that can be reduced (EAC) (Fig. 3). Surprisingly, the EAC values were larger for all FA and HA extracts obtained under anoxic extraction conditions than under oxic conditions (Fig. 3). The higher EAC under anoxic conditions suggests the presence of more functional groups that can be reduced in FA and HA extracted in the absence of oxygen, meaning that the additional number of organic compounds that were extracted under anoxic conditions compared to oxic conditions contain more oxidized functional groups.
3.3 Effects of different organic matter extracts on rate and extent of microbial ferrihydrite reduction and mineral transformation during reduction
To determine the effects of water-extractable OM, FA, and HA extracts on microbial Fe(III) reduction, the Fe(III) mineral ferrihydrite was incubated with the Fe(III)-reducing bacterium Shewanella oneidensis MR-1 in experiments amended with our extracts, and total Fe(II) concentration was monitored over time (Fig. 4). The highest initial microbial Fe(III) reduction rates were determined as shown in the Supplement (Fig. S3). The presence of AQDS stimulated ferrihydrite reduction to Fe(II) with a maximum reduction rate of 3.12±0.38 mmol Fe(II) d−1 compared to experiments without electron shuttle with a rate of 0.79±0.31 mmol Fe(II) d−1 (Figs. 4a, S3). The observed decrease in total Fe(II) after 5 d of incubation (from 14.67 to 6.87 mM) in the AQDS-amended setup was caused by Fe(II) loss due to sorption of Fe(II) or precipitation of Fe(II) (e.g., as Fe(II) phosphate mineral due to the presence of phosphate buffer) at the wall of the glass bottles (Fig. S1). After the addition of oxically and anoxically water-extractable OM (Fig. 4d), Fe(III) was reduced at maximum rates of 1.53±0.20 mmol Fe(II) d−1 and 2.07±0.43 mmol Fe (II) d−1, respectively, suggesting higher reduction rates than without any electron shuttle (0.79±0.31 mmol Fe(II) d−1).
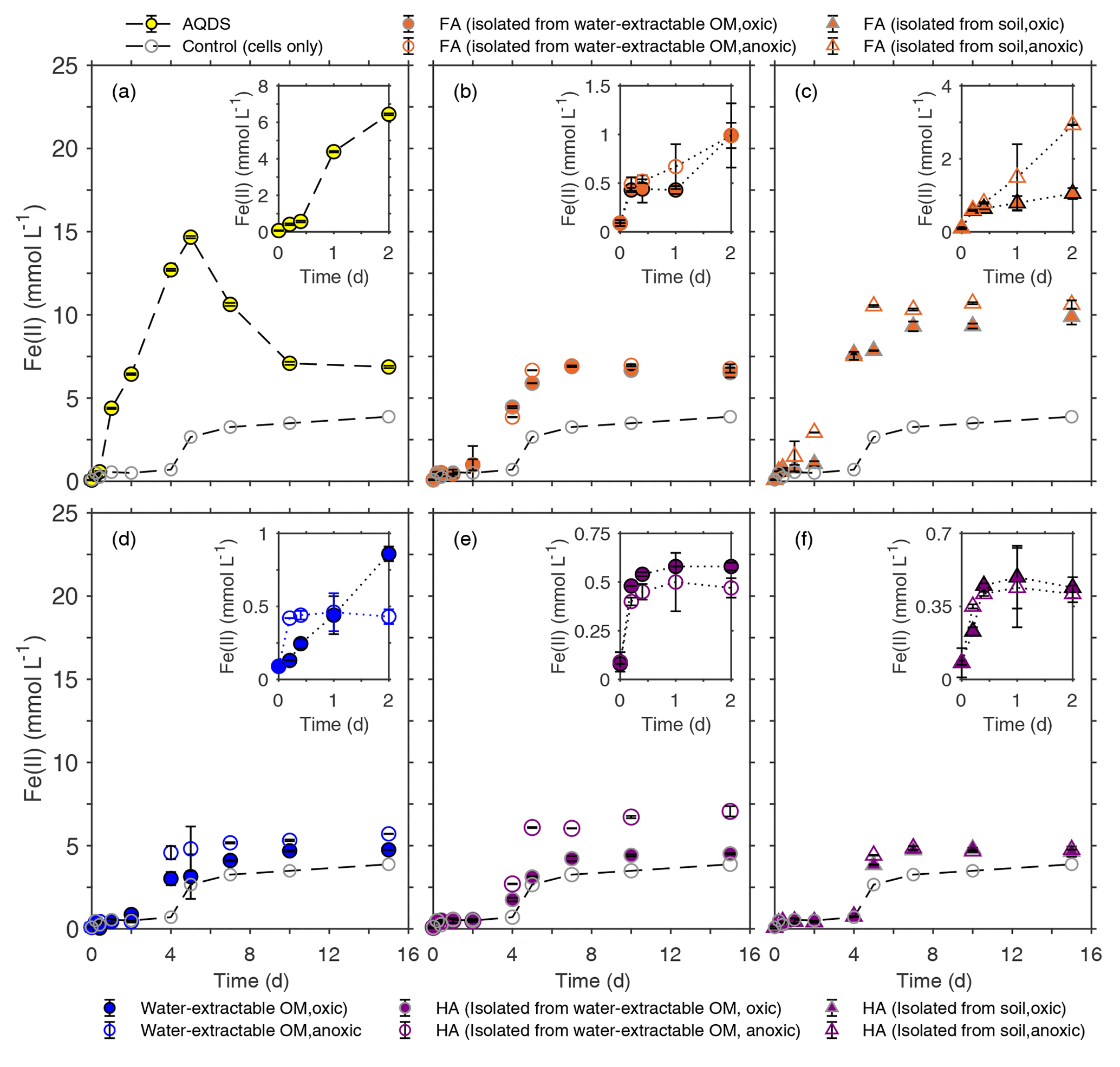
Figure 4Microbial reduction of ferrihydrite (15 mM) by S. oneidensis MR-1 (107 cells mL−1) in the presence of 15 mmol L−1 lactate as electron donor and 50 mg C L−1 FA (b, c), water-extractable OM (d), and HA (e, f) compared to 100 µmol L−1 AQDS (a) presented as formation of total Fe(II) over time. The inserts in panels (a)–(f) show the data points for the first 2 d of incubation. All experiments were incubated in airtight 100 mL glass serum bottles flushed with N2 at 30 ∘C in the dark. Control samples were incubated at the same condition in the absence of electron shuttles (ferrihydrite, lactate, and cells only). Error bars represent standard deviations of triplicate bottles.
When comparing Fe(III) reduction in the presence of the different HA extracts (Fig. 4e, f), we found that amendment with HA isolated from soil under anoxic conditions showed the fastest reduction rate (1.83±0.03 mmol Fe(II) d−1) followed by HA isolated from anoxically water-extractable OM (1.70±0.25 mmol Fe(II) d−1) and HA isolated from soil oxically (1.55±0.08 mmol Fe(II) d−1). The reduction rate of the experiment amended with HA isolated oxically from the water-extractable OM was 0.82±0.27, showing a slight stimulation effect compared to the setup without electron shuttle (0.79±0.31 mmol Fe(II) d−1). The addition of FA increased Fe(III) reduction rates significantly in all cases (Fig. 4b, c). In the presence of FA isolated oxically and anoxically from water-extractable OM, the fastest rates were 2.03±0.54 and 2.22±0.36 mmol Fe(II) d−1, respectively. After addition of FA isolated from soil under oxic and anoxic conditions, the maximum reduction rates were even faster with 2.31±0.15 and 3.05±0.07 mmol Fe(II) d−1. Control samples with only OM and ferrihydrite (without bacteria) did not show any ferrihydrite reduction (Fig. S2).
In addition to differences in reduction rates depending on the identity of the added organic extract, we also found differences in reduction extents. In most cases, the reduction extent was higher in the presence of OM compared to OM-free experiments (3.87 mM of Fe(II) after 15 d). Specifically, experiments amended with FA showed higher microbial Fe(III) reduction extents than with HA. After 25 d of incubation, experiments with FA extracted chemically from soil under anoxic conditions reduced 10.87 mmol L−1 Fe(III) to Fe(II), while the maximum Fe(III) reduction extent in the presence of added HA (chemically isolated from anoxically water-extracted SOM) was about 7.08 mmol L−1 Fe(II) (Fig. 4c and e).
Since the used OM extracts contained some Fe(II) and Fe(III), we evaluated the contribution of these ions to the observed Fe(III) reduction (Table S3). First, the Fe(II) present in the water-extractable OM, FA, and HA ranged from 7.2 (FA isolated from water-extractable OM, oxic) to 79.2 µmol L−1 (water-extractable OM, anoxic) (Table S3) and made up between 1 % and 17.6 % of the measured Fe(II) concentration after 30 min of incubation. With the increase in Fe(II) concentration over time, the percentage of Fe(II) present in the extracts to the measured Fe(II) concentration decreased to less than 0.1 % and is therefore negligible. Second, the influence of Fe(III) initially present in the water-extracted SOM, FA, and HA (Table S3) can be neglected as well, because the Fe(III) concentration of the extracted organic matter fractions ranged from 8.7 to 43.9 µmol L−1 (Table S3), but the ferrihydrite concentration used in the experiments was 15 mmol L−1.
4.1 Effects of the presence of oxygen on the amount and properties of SOM extracts
The presence and absence of oxygen impacted the amount of water-extractable OM. Under anoxic conditions, water at neutral pH extracted about 6.7 times more organic carbon than under oxic conditions (Table 1). The presence of Fe(II) at the end of extraction in all anoxic extracts suggested that the higher amount of extracted OM is probably related to microbial Fe(III) mineral reduction and the release of mineral-bound OM during mineral dissolution. A correlation between the dissolved organic carbon (DOC) concentration and the amount of Fe(II) in pore water was reported before for sediment samples that were incubated in the dark under anoxic conditions for 14 d (Dadi et al., 2017). In situ monitoring of the DOC flux in pore water of marine sediment or freshwater wetland also suggested an increase in DOC with increasing microbial iron(III) mineral reduction (Burdige et al., 1992, 1999; Chin et al., 1998). Other studies also suggested an increase in DOC under anoxic conditions due to the microbial iron(III) mineral reduction and dissolution and the concomitant release of organic carbon (OC) that was co-precipitated with and adsorbed to the iron(III) minerals (Gu et al., 1994; Riedel et al., 2013; Shimizu et al., 2013).
In addition to differences in the amount of extracted OM, the presence or absence of oxygen also influenced the aromaticity of the extracted SOM, as shown by the SUVA254 values (Table 1) and 13C-NMR data (Fig. 2). Water-extractable OM extracted under anoxic conditions showed a higher aromaticity, suggesting that the additional organic matter mobilized by reductive dissolution of iron minerals possesses a higher degree of aromaticity. This is in line with findings described by other studies (Gu et al., 1994; Lv et al., 2016; Avneri-Katz et al., 2017; Coward et al., 2019). Kothawala et al. (2012) incubated oxically extracted soil solution with soils with different mineral composition. SUVA and fluorescence index analysis of the remaining non-sorbed organic matter showed that regardless of the soil type, the aromatic functional groups were preferentially adsorbed to the soil minerals.
4.2 Effect of extraction pH on the amount and properties of extracted organic matter
The practice of extracting and isolating HA and FA using NaOH and HCl under anoxic conditions has been the established standard protocol (IHSS, 2019). As early as 1972, Swift and Posner (1972) showed that by incubating a peat HA with 1 M NaOH under oxic conditions for 30 d, more than half of the HA was degraded to low-molecular-weight molecules and amino acid N was lost from the HA. Later studies also reported the hydrolysis of esters in natural organic matter (NOM) to carboxylic acid groups when exposing NOM to NaOH under oxic conditions (Ritchie and Perdue, 2008). Consistent with previous studies, our SUVA254 (Table 1), 13C-NMR (Fig. 2), and EEC (Fig. 3) results showed that FA and HA isolated from soil under oxic conditions had lower aromaticity and EEC compared to the FA and HA isolated from soil under anoxic conditions. This indicates that degradation of aromatic structures and functional groups in the OM to smaller molecules occurs in the presence of O2 under higher-pH conditions.
However, we found that even under anoxic conditions, the chemical extraction extracted up to 100 times more carbon than the water extraction at a neutral pH (Table 1), consistent with previous studies (Aiken, 1985). This higher extraction efficiency at a high pH could be due to the deprotonation of carboxyl and phenol functional groups leading to both higher aqueous solubility and electrostatic repulsion of OM from negatively charged soil minerals (Kleber et al., 2015) or due to the hydrolysis of plant material and the formation of smaller oligo- and monomers (Sparks, 2003). Not only the amount of C extracted but also the properties of the extracted FA and HA are affected by the chemical extraction under anoxic conditions. Our results indicate that the HA isolated from soil has higher aromaticity than water-extractable OM under anoxic extraction conditions. On the one hand, the higher aromaticity in HA isolated from soil can probably be explained by the extra amount of C extracted from soil by the chemical extraction method. On the other hand, this cannot be the only explanation, since the HA isolated from the water-extractable OM also had higher aromaticity than the water-extractable OM itself. This suggests the formation of aromatic functional groups during the extraction with NaOH under anoxic conditions by condensation reactions between amino acids, aldehydes, and hydroxyl-and catechol-containing molecules. Such condensation reactions could result in larger molecules with a higher degree of aromaticity (Gieseking, 1975; Golchin et al., 1994; Kappler and Brune, 1999; Kappler and Haderlein, 2003). A recent study comparing OM extraction from a freshwater sediment using water (acidified to pH 2 with 1 M HCl) with an extraction using 0.1 M sodium pyrophosphate (pH 10) and 0.5 M NaOH (pH 12) also revealed a higher aromaticity in the alkali-extracted OM (Fox et al., 2017). Using Fourier-transform infrared spectroscopy (FTIR) and electrospray ionization Fourier-transform ion cyclotron resonance mass spectrometry (ESI-FTICR-MS), these authors showed that OM extracted by sodium pyrophosphate and NaOH had more condensed aromatic compounds.
4.3 Electron-exchange capacity (EEC) of soil extracts determines their ability to stimulate microbial Fe(III) reduction
Our data showed that the rates of microbial ferrihydrite reduction differed in the presence of different OM extracts. The observed differences in Fe(III) reduction rates can either be a result of the differences in OM redox activity (e.g., number and redox potentials of redox-active functional groups) and the resulting function of the OM as an electron shuttle or due to different secondary mineral phases that can form during ferrihydrite reduction. However, a previous microbial Fe(III) mineral reduction study of ferrihydrite (5 mM Fe(III)) in the presence of 50 mg C L−1 OM, 0.8 mM phosphate buffer, and 2×105 cells mL−1 Shewanella oneidensis MR-1 showed no goethite or magnetite (based on 57Fe-Mössbauer and X-ray powder diffraction (XRD) analysis) but vivianite as the major mineral phase produced (Amstaetter et al., 2012). The transformation of ferrihydrite to vivianite instead of goethite or magnetite in the presence of phosphate buffer was also reported in other studies using similar concentrations of OM, buffer, cells, and ferrihydrite (Chen et al., 2003a; Piepenbrock et al., 2011; Shimizu et al., 2013). The formation of more crystalline secondary mineral phases such as goethite was only observed during ferrihydrite reduction in the absence of phosphate (Hansel et al., 2003; Borch et al., 2007). Abiotic experiments showed that phosphate inhibits the transformation of ferrihydrite to magnetite or goethite by blocking of surface sites of ferrihydrite, therefore preventing the sorption of the produced Fe(II) on the Fe(III) mineral, and thus lowering the number of surface sites where conversion of ferrihydrite to magnetite or goethite can take place (Galvez et al., 1999). Therefore, the transformation of ferrihydrite to magnetite or goethite is not expected to happen in our experiments and the following discussion will focus on the influence of the redox activity of the extracted OM on the rate and extent of the microbial Fe(III) reduction.
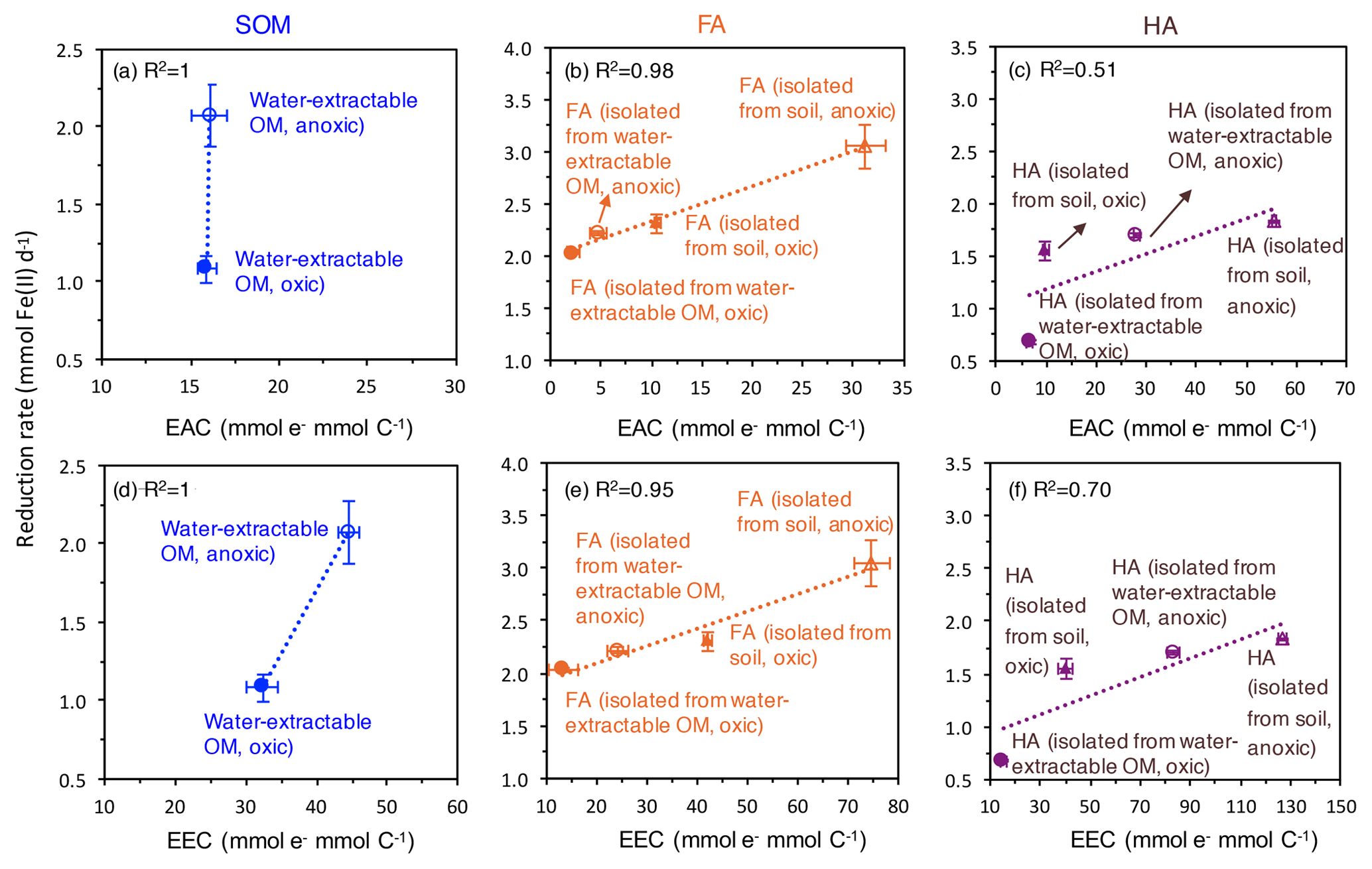
Figure 5Correlation of the electron-exchange capacity (EEC) (d, e, f) and electron-accepting capacity (EAC) (a, b, c) with the fastest microbial ferrihydrite reduction rates (Shewanella oneidensis MR-1) in the presence of oxically and anoxically prepared SOM (a), FA (b), and HA (c). EEC and EAC values are replotted from Fig. 2. Please note that the EEC and EAC values were determined by the electrochemical method described in the Methods section and represent the contribution of both redox-active organic functional groups such as quinones and the redox-active Fe ions in the SOM, FA, and HA extracts. Horizontal error bars represent standard deviations of the measured EEC values; vertical error bars are standard deviations of the reduction rates calculated as shown in Fig. S4.
As measures for the redox activity of the different extracted OM fractions, we determined their potential for accepting electrons (EAC) and for accepting and donating electrons (EEC). Correlating the EAC values of our different OM extracts and the maximum microbial ferrihydrite reduction rates showed that the higher the EAC values of the extracted OM, FA, and HA, the faster the microbial Fe(III) reduction rates are (Fig. 5). As shown before (Aeschbacher et al., 2012), (hydro)quinone functional groups contribute mainly to the measured EAC values in OM, and these quinone moieties are thought to be the major functional group responsible for electron transfer between Fe(III)-reducing bacteria and Fe(III) minerals during electron shuttling. Scott et al. (1998) reported a direct correlation between OM oxidation capacity and the stable free-radical content in the OM, stemming from semiquinone radicals (Lovley et al., 1996; Scott et al., 1998). However, we also found a correlation between EEC values and the maximum microbial ferrihydrite reduction rates in the presence of water-extractable OM, HA, and FA. Since higher EEC values reflect higher contents of aromatic and polycondensed aromatic compounds in the OM (Aeschbacher et al., 2012), our results also indicate that, apart from quinones, other aromatic functional groups were also involved in the microbial Fe(III) reduction with OM as electron shuttles, and these functional groups also influence the electron transfer efficiency between the Fe(III)-reducing bacteria, the OM, and the Fe(III) minerals. Support for the participation of non-quinone groups in such OM electron transfer studies also comes from previous analyses of redox properties and stable free-radical concentrations in OM (Struyk and Sposito, 2001; Chen et al., 2003a).
Faster Fe(III) mineral reduction rates in the presence of more aromatic functional groups (including quinones) were demonstrated previously in experiments with increasing concentrations of AQDS or HA (Jiang and Kappler, 2008; Wolf et al., 2009; Glasser et al., 2017). It was suggested that the microbial turnover of substrate (lactate as electron source) is limited by the availability of the electron acceptor, i.e., either by the Fe(III) in the absence of shuttles or by the OM when OM serves as an electron-shuttling compound (Jiang and Kappler, 2008; Poggenburg et al., 2018). Thus, with the same concentration of OM electron shuttle, the OM with more redox-active functional groups can accept more electrons per time from the microorganisms, therefore resulting in higher Fe(III) reduction rates. Additionally, when more quinone or other redox-active functional groups are present per shuttle molecule, the distance between redox-active functional groups is smaller; therefore, electron transfer within the shuttle molecule and between the shuttle molecules can occur faster, thus further increasing the electron transfer rate from the microbial cells to the shuttle molecules and further to the Fe(III) minerals (Boyd et al., 2015; Glasser et al., 2017). The different types and proportions of functional groups in the different OM extracts may also influence their adsorption onto the ferrihydrite surface and therefore also impact the rates of microbial ferrihydrite reduction amended with different OM. However, due to the high concentration of lactate and HEPES buffer in our experiment, we could not quantify the amount of adsorbed OM vs. dissolved OM. It has to be noted, however, that in our extracted OM (Fig. 2) different amounts of redox-active Fe ions were present and that the redox-active OM-bound Fe can potentially also influence the rates of Fe(III) mineral reduction. The OM-bound Fe(III) can also be reduced by the Fe(III)-reducing bacteria or by reduced organic functional groups in the OM to Fe(II), which can then transfer electrons further to the ferrihydrite. The OM-bound Fe is subsequently reoxidized to Fe(III) and therefore contributes to electron shuttling between Fe(III)-reducing bacteria and ferrihydrite.
In addition to the differences in the reduction rates, we also observed that the extent of ferrihydrite reduction was influenced by the presence of different OM. Specifically, amendments with HA lead to lower extents of Fe(III) reduction than FA amendments. This difference could be caused by the higher content of aromatic functional groups in HA than in FA and the resulting differences in sorption properties. OM with higher aromaticity and higher molecular weight was shown to have a higher adsorption affinity to ferrihydrite (Lv et al., 2016; Coward et al., 2019). Since our HAs extracted under all conditions were more aromatic than the FA, the HAs were probably preferentially adsorbed to ferrihydrite. On the one hand, the sorbed HA can block surface sites on the minerals and restrict the accessibility for bacteria. On the other hand, HA adsorption changes the net surface charge of ferrihydrite from positive to (partially) negative and thus leads to repulsion of negatively charged cells (Aeschbacher et al., 2012).
In summary, our results clearly show that the extraction method determines the concentration of redox-active (aromatic) functional groups, and the EEC of the soil extracts and the EEC is a key factor for the electron-shuttling capacity of soil extracts in microbial Fe(III) mineral reduction. Therefore, it has to be carefully decided which SOM extraction method to apply and which SOM fraction to use in biogeochemical experiments to obtain soil extracts that can represent natural SOM. Representative SOM is necessary to obtain meaningful results that will prevent overestimating the reactivity of SOM in redox processes in the environment. Based on our experimental results we suggest that, firstly, the NaOH extraction method should be avoided in general because it alters the chemical and redox properties of SOM. Additionally, soil pH values typically range from 3.5 to 8.5 (Sparks, 2003); therefore the organic matter that is soluble only at pH > 12 will not be dissolved under in situ soil conditions and might react differently in biogeochemical processes compared to solid-phase soil OM (Roden et al., 2010; Kappler et al., 2014). Secondly, when extracting SOM with water at neutral pH, the redox milieu (oxic or anoxic) during extraction needs to be carefully controlled. When targeting oxic environmental systems with the goal of obtaining relevant OM matter that participates in biogeochemical processes under such redox conditions, short extraction times (<24 h), small batches, aeration, and thorough stirring are recommended for the OM extraction. Thus, anoxic conditions during the OM extraction that would lead to reductive dissolution of iron minerals with concomitant mobilization of OM that would not be available under oxic conditions should be avoided (in the absence of microbial Fe(III) reduction). However, in the case that target environmental systems undergo redox fluctuations or even permanent reducing conditions, yielding anoxic conditions with microbial Fe(III) mineral reduction during the OM extraction is appropriate.
Raw data of all results presented in this study are available at https://doi.org/10.1594/PANGAEA.904416 (Bai et al., 2019).
The Supplement includes the statistical analysis of the specific UV absorbance at 254 nm (SUVA254) data, the 13C-NMR analysis, the Fe(II) and Fe(III) content and their contribution to the electron-exchange capacity (EEC) of the OM extracts, the statistical analysis of the EEC data, the picture showing the sorption and precipitation of Fe(II) on the wall of glass bottles of the microbial Fe(III) reduction experiments containing AQDS, the results of the abiotic reduction of Fe(III) minerals with the addition of OM extracts, and the calculation of the rates of microbial Fe(III) reduction amended with different OM extracts. The supplement related to this article is available online at: https://doi.org/10.5194/bg-17-683-2020-supplement.
YB, AK, and SH designed the experiment. YB conducted the experiments and analyzed the data of OM extraction, and microbial Fe(III) reduction together with AK. ES performed and analyzed data of the electron chemical analysis and helped with the fluorescence spectra. HK conducted the NMR analysis and processed the data. YB and AK prepared the paper, with great help from SH.
The authors declare that they have no conflict of interest.
We acknowledge Thomas Scholten (University of Tübingen) for his help with soil sampling. We thank Ellen Röhm, Lars Grimm, and Franziska Schaedler for technical support and Alison Enright, Zhe Zhou, and Zhen Yang for their help on improving the manuscript.
This research has been supported by the Deutsche Forschungsgemeinschaft (grant no. KA 1736/37-1).
This open-access publication was funded
by the University of Tübingen.
This paper was edited by Denise Akob and reviewed by two anonymous referees.
Abelmann, K., Kleineidam, S., Knicker, H., Grathwohl, P., and Koegel-Knabner, I.: Sorption of HOC in soils with carbonaceous contamination: Influence of organic-matter composition, J. Plant Nutr. Soil Sc., 168, 293–306, https://doi.org/10.1002/jpln.200421622, 2005.
Achard, F. K.: Chemische Untersuchung des Torfs, Chem. Ann. Freunde Naturlehre, Arzneigel. Hausalt. Manufact, 2, 391–403, 1786.
Aeschbacher, M., Sander, M., and Schwarzenbach, R. P.: Novel electrochemical approach to assess the redox properties of humic substances, Environ. Sci. Technol., 44, 87–93, https://doi.org/10.1021/es902627p, 2010.
Aeschbacher, M., Graf, C., Schwarzenbach, R. P., and Sander, M.: Antioxidant properties of humic substances, Environ. Sci. Technol., 46, 4916–4925, https://doi.org/10.1021/es300039h, 2012.
Aiken, G. R.: Humic substances in soil, sediment, and water: geochemistry, isolation, and characterization, Wiley, New York, 1985.
Amstaetter, K., Borch, T., and Kappler, A.: Influence of humic acid imposed changes of ferrihydrite aggregation on microbial Fe(III) reduction, Geochim. Cosmochim. Ac., 85, 326–341, https://doi.org/10.1016/j.gca.2012.02.003, 2012.
Avneri-Katz, S., Young, R. B., McKenna, A. M., Chen, H., Corilo, Y. E., Polubesova, T., Borch, T., and Chefetz, B.: Adsorptive fractionation of dissolved organic matter (DOM) by mineral soil: Macroscale approach and molecular insight, Org. Geochem., 103, 113–124, https://doi.org/10.1016/j.orggeochem.2016.11.004, 2017.
Bai, Y., Subdiaga, E., Haderlein, S. B., Knicker, H., and Kappler, A.: Supplementary data for: Effects of extraction conditions on the redox properties of soil organic matter (SOM) and its ability to stimulate microbial iron(III) mineral reduction by electron shuttling, PANGAEA, https://doi.org/10.1594/PANGAEA.904416, 2019.
Bauer, I. and Kappler, A.: Rates and extent of reduction of Fe(III) compounds and O2 by humic substances, Environ. Sci. Technol., 43, 4902–4908, https://doi.org/10.1021/es900179s, 2009.
Borch, T., Masue, Y., Kukkadapu, R. K., and Fendorf, S.: Phosphate imposed limitations on biological reduction and alteration of ferrihydrite, Environ. Sci. Technol., 41, 166–172, https://doi.org/10.1021/es060695p, 2007.
Boyd, D. A., Snider, R. M., Erickson, J. S., Roy, J. N., Strycharz-Glaven, S. M., and Tender, L. M.: Theory of redox conduction and the measurement of electron transport rates through electrochemically active biofilms, edited by: Beyenal, H. and Babauta, J., Wiley, New Jersey, 177–210, https://doi.org/10.1002/9781119097426.ch6, 2015.
Burdige, D. J., Alperin, M. J., Homstead, J., and Martens, C. S.: The role of Benthic fluxes of dissolved organic-carbon in oceanic and sedimentray carbon cycling, Geophys. Res., 19, 1851–1854, https://doi.org/10.1029/92GL02159, 1992.
Burdige, D. J., Berelson, W. M., Coale, K. H., McManus, J., and Johnson, K. S.: Fluxes of dissolved organic carbon from California continental margin sediments, Geochim. Cosmochim. Ac., 63, 1507–1515, https://doi.org/10.1016/S0016-7037(99)00066-6, 1999.
Chen, J., Gu, B. H., Royer, R. A., and Burgos, W. D.: The roles of natural organic matter in chemical and microbial reduction of ferric iron, Sci. Total Environ., 307, 167–178, https://doi.org/10.1016/S0048-9697(02)00538-7, 2003a.
Chin, Y. P., Traina, S. J., Swank, C. R., and Backhus, D.: Abundance and properties of dissolved organic matter in pore waters of a freshwater wetland, Limnol. Oceanogr., 43, 1287–1296, https://doi.org/10.4319/lo.1998.43.6.1287, 1998.
Coward, E. K., Ohno, T., and Sparks, D. L.: Direct evidence for temporal molecular fractionation of dissolved organic matter at the iron oxyhydroxide interface, Environ. Sci. Technol., 53, 642–650, https://doi.org/10.1021/acs.est.8b04687, 2019.
Dadi, T., Harir, M., Hertkorn, N., Koschorreck, M., Schmitt-Kopplin, P., and Herzsprung, P.: Redox conditions affect dissolved organic carbon quality in stratified freshwaters, Environ. Sci. Technol., 51, 13705–13713, https://doi.org/10.1021/acs.est.7b04194, 2017.
Engebretson, R. R. and Von Wandruszka, R.: Effects of humic acid purification on interactions with hydrophobic organic matter: Evidence from fluorescence behavior, Environ. Sci. Technol., 33, 4299–4303, https://doi.org/10.1021/es990386h, 1999.
Fischlin, A. M., Price, J., Leemans, R., Gopal, B., Turley, C., Rounsevell, M. D. A., Dube, P., Tarazona, J., and Velichko, A. A.: Ecosystems, their properties, goods and services, in: Climate Change 2007: Impacts, Adaptation and Vulnerability. Contribution of Working Group II on the Fourth Assessment Respot of the intergovernmental Panel on Climate Change, edited by: Parry, M. L., Canziani, O. F., Palutikof, J. P., van der Linden, P. J., and Hanson, C. E., Cambridge University Press, Cambridge, UK and Newyork, USA, 211–272, 2007.
Fox, P. M., Nico, P. S., Tfaily, M. M., Heckman, K., and Davis, J. A.: Characterization of natural organic matter in low-carbon sediments: Extraction and analytical approaches, Org. Geochem., 114, 12–22, https://doi.org/10.1016/j.orggeochem.2017.08.009, 2017.
Galvez, N., Barron, V., and Torrent, J.: Effect of phosphate on the crystallization of hematite, goethite, and lepidocrocite from ferrihydrite, Clay. Clay Miner., 47, 304–311, https://doi.org/10.1346/CCMN.1999.0470306, 1999.
Gieseking, J. E.: Soil components, Vol. 1, Organic components, Springer, Heidelberg, West Germany, 1975.
Glasser, N. R., Saunders, S. H., and Newman, D. K.: The colorful world of extracellular electron shuttles, Annu. Rev. Microbiol., 71, 731–751, https://doi.org/10.1146/annurev-micro-090816-093913, 2017.
Golchin, A., Oades, J. M., Skjemstad, J. O., and Clarke, P.: Soil structure and carbon cycling, Aust. J. Soil. Res., 32, 1043–1068, https://doi.org/10.1071/Sr9941043, 1994.
Gu, B. H., Schmitt, J., Chen, Z. H., Liang, L. Y., and McCarthy, J. F.: Adsorption and desorption of natural organic-matter on iron-oxide – Mechanisms and models, Environ. Sci. Technol., 28, 38–46, https://doi.org/10.1021/es00050a007, 1994.
Hansel, C. M., Benner, S. G., Neiss, J., Dohnalkova, A., Kukkadapu, R. K., and Fendorf, S.: Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow, Geochim. Cosmochim. Ac., 67, 2977–2992, https://doi.org/10.1016/S0016-7037(03)00276-X, 2003.
Hegler, F., Posth, N. R., Jiang, J., and Kappler, A.: Physiology of phototrophic iron(II)-oxidizing bacteria: implications for modern and ancient environments, FEMS Microbiol. Ecol., 66, 250–260, https://doi.org/10.1111/j.1574-6941.2008.00592.x, 2008.
IHSS: Isolation of IHSS samples, available at: http://humic-substances.org/isolation-of-ihss-samples/, last access: 29 July 2019.
Jiang, J. and Kappler, A.: Kinetics of microbial and chemical reduction of humic substances: Implications for electron shuttling, Environ. Sci. Technol., 42, 3563–3569, https://doi.org/10.1021/es7023803, 2008.
Kappler, A. and Brune, A.: Influence of gut alkalinity and oxygen status on mobilization and size-class distribution of humic acids in the hindgut of soil-feeding termites, Appl. Soil. Ecol., 13, 219–229, https://doi.org/10.1016/S0929-1393(99)00035-9, 1999.
Kappler, A. and Haderlein, S. B.: Natural organic matter as reductant for chlorinated aliphatic pollutants, Environ. Sci. Technol., 37, 2714–2719, https://doi.org/10.1021/es0201808, 2003.
Kappler, A., Benz, M., Schink, B., and Brune, A.: Electron shuttling via humic acids in microbial iron(III) reduction in a freshwater sediment, FEMS Microbiol. Ecol., 47, 85–92, https://doi.org/10.1016/S0168-6496(03)00245-9, 2004.
Kappler, A., Wuestner, M. L., Ruecker, A., Harter, J., Halama, M., and Behrens, S.: Biochar as an electron shuttle between bacteria and Fe(III) minerals, Environ. Sci. Tech. Let., 1, 339–344, https://doi.org/10.1021/ez5002209, 2014.
Kleber, M. and Lehmann, J.: Humic substances extracted by alkali are invalid proxies for the dynamics and functions of organic matter in terrestrial and aquatic ecosystems, J. Environ. Qual., 48, 207–216, https://doi.org/10.2134/jeq2019.01.0036, 2019.
Kleber, M., Eusterhues, K., Keiluweit, M., Mikutta, C., Mikutta, R., and Nico, P. S.: Mineral-organic associations: Formation, properties, and relevance in soil environments, Adv. Agron., 130, 1–140, https://doi.org/10.1016/bs.agron.2014.10.005, 2015.
Klupfel, L., Piepenbrock, A., Kappler, A., and Sander, M.: Humic substances as fully regenerable electron acceptors in recurrently anoxic environments, Nat. Geosci., 7, 195–200, https://doi.org/10.1038/Ngeo2084, 2014.
Korshin, G. V., Li, C. W., and Benjamin, M. M.: Monitoring the properties of natural organic matter through UV spectroscopy: A consistent theory, Water Res., 31, 1787–1795, https://doi.org/10.1016/S0043-1354(97)00006-7, 1997.
Kothawala, D. N., Roehm, C., Blodau, C., and Moore, T. R.: Selective adsorption of dissolved organic matter to mineral soils, Geoderma, 189, 334–342, https://doi.org/10.1016/j.geoderma.2012.07.001, 2012.
Lal, R.: Soil carbon sequestration impacts on global climate change and food security, Science, 304, 1623–1627, https://doi.org/10.1126/science.1097396, 2004.
Lehmann, J. and Kleber, M.: The contentious nature of soil organic matter, Nature, 528, 60–68, https://doi.org/10.1038/nature16069, 2015.
Lies, D. P., Hernandez, M. E., Kappler, A., Mielke, R. E., Gralnick, J. A., and Newman, D. K.: Shewanella oneidensis MR-1 uses overlapping pathways for iron reduction at a distance and by direct contact under conditions relevant for biofilms, Appl. Environ. Microb., 71, 4414–4426, https://doi.org/10.1128/AEM.71.8.4414-4426.2005, 2005.
Lovley, D. R. and Blunt-Harris, E. L.: Role of humic-bound iron as an electron transfer agent in dissimilatory Fe(III) reduction, Appl. Environ. Microb., 65, 4252–4254, 1999.
Lovley, D. R., Coates, J. D., BluntHarris, E. L., Phillips, E. J. P., and Woodward, J. C.: Humic substances as electron acceptors for microbial respiration, Nature, 382, 445–448, 1996.
Lovley, D. R., Fraga, J. L., Blunt-Harris, E. L., Hayes, L. A., Phillips, E. J. P., and Coates, J. D.: Humic substances as a mediator for microbially catalyzed metal reduction, Acta. Hydroch. Hydrob., 26, 152–157, https://doi.org/10.1002/(Sici)1521-401x(199805)26:3<152::Aid-Aheh152>3.0.Co;2-D, 1998.
Lv, J. T., Zhang, S. Z., Wang, S. S., Luo, L., Cao, D., and Christie, P.: Molecular-scale investigation with ESI-FT-ICR-MS on fractionation of dissolved organic matter induced by adsorption on iron oxyhydroxides, Environ. Sci. Technol., 50, 2328–2336, https://doi.org/10.1021/acs.est.5b04996, 2016.
Marin-Spiotta, E., Gruley, K. E., Crawford, J., Atkinson, E. E., Miesel, J. R., Greene, S., Cardona-Correa, C., and Spencer, R. G. M.: Paradigm shifts in soil organic matter research affect interpretations of aquatic carbon cycling: transcending disciplinary and ecosystem boundaries, Biogeochemistry, 117, 279–297, https://doi.org/10.1007/s10533-013-9949-7, 2014.
Maurer, F., Christi, I., and Kretzschmar, R.: Reduction and reoxidation of humic acid: Influence on spectroscopic properties and proton binding, Environ. Sci. Technol., 44, 5787–5792, https://doi.org/10.1021/es100594t, 2010.
Murphy, B. W.: Soil organic matter and soil function – review of the literature and underlying data: effects of soil organic matter on functional soil properties, Department of Environment, Canberra, Australia, 155 pp., 2014.
Piccolo, A.: Characteristics of soil humic extracts obtained by some organic and inorganic solvents and purified by HCl-HF treatment, Soil Sci., 146, 418–426, https://doi.org/10.1097/00010694-198812000-00003, 1988.
Piepenbrock, A., Dippon, U., Porsch, K., Appel, E., and Kappler, A.: Dependence of microbial magnetite formation on humic substance and ferrihydrite concentrations, Geochim. Cosmochim. Ac., 75, 6844–6858, https://doi.org/10.1016/j.gca.2011.09.007, 2011.
Piepenbrock, A., Schroder, C., and Kappler, A.: Electron transfer from humic substances to biogenic and abiogenic Fe(III) oxyhydroxide minerals, Environ. Sci. Technol., 48, 1656–1664, https://doi.org/10.1021/es404497h, 2014.
Poggenburg, C., Mikutta, R., Schippers, A., Dohrmann, R., and Guggenberger, G.: Impact of natural organic matter coatings on the microbial reduction of iron oxides, Geochim. Cosmochim. Ac., 224, 223–248, https://doi.org/10.1016/j.gca.2018.01.004, 2018.
Riedel, T., Zak, D., Biester, H., and Dittmar, T.: Iron traps terrestrially derived dissolved organic matter at redox interfaces, P. Natl. Acad. Sci. USA, 110, 10101–10105, https://doi.org/10.1073/pnas.1221487110, 2013.
Ritchie, J. D. and Perdue, E. M.: Analytical constraints on acidic functional groups in humic substances, Org. Geochem., 39, 783–799, https://doi.org/10.1016/j.orggeochem.2008.03.003, 2008.
Roden, E. E., Kappler, A., Bauer, I., Jiang, J., Paul, A., Stoesser, R., Konishi, H., and Xu, H. F.: Extracellular electron transfer through microbial reduction of solid-phase humic substances, Nat. Geosci., 3, 417–421, https://doi.org/10.1038/Ngeo870, 2010.
Schad, P., van Huysteen, C., and Micheli, E. (Eds.): World reference base for soil rescouces 2014 – International soil classification system for naming soils and creating legends for soil maps, FAO, Rome, Italy, 2014.
Scott, D. T., McKnight, D. M., Blunt-Harris, E. L., Kolesar, S. E., and Lovley, D. R.: Quinone moieties act as electron acceptors in the reduction of humic substances by humics-reducing microorganisms, Environ. Sci. Technol., 32, 2984–2989, https://doi.org/10.1021/es980272q, 1998.
Shimizu, M., Zhou, J., Schröder, C., Obst, M., Kappler, A., and Borch, T.: Dissimilatory reduction and transformation of ferrihydrite-humic acid coprecipitates, Environ. Sci. Technol., 47, 13375–13384, https://doi.org/10.1021/es402812j, 2013.
Sparks, D. L.: Environmental soil chemistry, Academic Press, Amsterdam, Boston, 2003.
Stookey, L. L.: Ferrozine – A new spectrophotometric reagent for iron, Anal. Chem., 42, 779–781, https://doi.org/10.1021/ac60289a016, 1970.
Struyk, Z. and Sposito, G.: Redox properties of standard humic acids, Geoderma, 102, 329–346, https://doi.org/10.1016/S0016-7061(01)00040-4, 2001.
Swift, R. S. and Posner, A. M.: Autoxidation of humic acid under alkaline conditions, J. Soil Sci., 23, 381–393, https://doi.org/10.1111/j.1365-2389.1972.tb01669.x, 1972.
van Bemmelen, J. M.: Die Absorptionsverbindungen und das Absorptionsvermögen der Ackererde, Landwirtschaftlichen Versychs-Stationen, 35, 2228–2233, https://doi.org/10.1007/BF01381567, 1888.
Weishaar, J. L., Aiken, G. R., Bergamaschi, B. A., Fram, M. S., Fujii, R., and Mopper, K.: Evaluation of specific ultraviolet absorbance as an indicator of the chemical composition and reactivity of dissolved organic carbon, Environ. Sci. Technol., 37, 4702–4708, https://doi.org/10.1021/es030360x, 2003.
Wolf, M., Kappler, A., Jiang, J., and Meckenstock, R. U.: Effects of humic substances and quinones at low concentrations on ferrihydrite reduction by Geobacter metallireducens, Environ. Sci. Technol., 43, 5679–5685, https://doi.org/10.1021/es803647r, 2009.