the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Jan 2021
| 08 Jan 2021
Characterization of particle-associated and free-living bacterial and archaeal communities along the water columns of the South China Sea
Jiangtao Li
Lingyuan Gu
Shijie Bai
Jie Wang
Lei Su
Bingbing Wei
Li Zhang
Jiasong Fang
There is a growing recognition of the role of particle-attached (PA) and free-living (FL) microorganisms in marine carbon cycle. However, current understanding of PA and FL microbial communities is largely focused on those in the upper photic zone, and relatively fewer studies have focused on microbial communities of the deep ocean. Moreover, archaeal populations receive even less attention. In this study, we determined bacterial and archaeal community structures of both the PA and FL assemblages at different depths, from the surface to the bathypelagic zone along two water column profiles in the South China Sea. Our results suggest that environmental parameters including depth, seawater age, salinity, particulate organic carbon (POC), dissolved organic carbon (DOC), dissolved oxygen (DO) and silicate play a role in structuring these microbial communities. Generally, the PA microbial communities had relatively low abundance and diversity compared with the FL microbial communities at most depths. Further microbial community analysis revealed that PA and FL fractions generally accommodate significantly divergent microbial compositions at each depth. The PA bacterial communities mainly comprise members of Alphaproteobacteria and Gammaproteobacteria, together with some from Planctomycetes and Deltaproteobacteria, while the FL bacterial lineages are also mostly distributed within Alphaproteobacteria and Gammaproteobacteria, along with other abundant members chiefly from Actinobacteria, Cyanobacteria, Bacteroidetes, Marinimicrobia and Deltaproteobacteria. Moreover, there was an obvious shifting in the dominant PA and FL bacterial compositions along the depth profiles from the surface to the bathypelagic deep. By contrast, both PA and FL archaeal communities dominantly consisted of euryarchaeotal Marine Group II (MGII) and thaumarchaeotal Nitrosopumilales, together with variable amounts of Marine Group III (MGIII), Methanosarcinales, Marine Benthic Group A (MBG-A) and Woesearchaeota. However, the pronounced distinction of archaeal community compositions between PA and FL fractions was observed at a finer taxonomic level. A high proportion of overlap of microbial compositions between PA and FL fractions implies that most microorganisms are potentially generalists with PA and FL dual lifestyles for versatile metabolic flexibility. In addition, microbial distribution along the depth profile indicates a potential vertical connectivity between the surface-specific microbial lineages and those in the deep ocean, likely through microbial attachment to sinking particles.
- Article
(3417 KB) - Full-text XML
-
Supplement
(2111 KB) - BibTeX
- EndNote





The sinking of particulate organic matter (POM) formed in the photic layer is a fundamental process that transports carbon and nutrient materials from the surface into the usually starved deep ocean, with a significant role in structuring the distributions and activities of marine microorganisms in the dark realm (Azam and Malfatti, 2007; Mestre et al., 2018; Suter et al., 2018). During sinking, the POM is generally colonized and concurrently decomposed by particle-attached (PA) prokaryotes, releasing dissolved organic matter (DOM) into ambient seawater, fueling the free-living (FL) microbes (Kiorboe and Jackson, 2001; Azam and Malfatti, 2007). It has been revealed that PA and FL microbial populations exhibit different taxonomic composition, physiology and metabolism, corresponding to their lifestyle and ecological behavior. For example, PA bacteria, compared to FL bacteria, are often larger in size (Alldredge et al., 1986; Zhang et al., 2007; Lauro et al., 2009) and metabolically more active (Karner and Herdl, 1992; Grossart et al., 2007). They often maintain higher levels of extracellular enzymes, adhesion proteins, and antagonistic compounds and are capable of degrading high-molecular-weight (HMW) organic compounds (Smith et al., 1992; Crump et al., 1998; Long and Azam, 2001; Mevel et al., 2008; Ganesh et al., 2014). An examination of microbial metagenomes suggests that there are notable differences between PA and FL assemblages in GC content, effective genome size, general taxonomic composition and functional gene categories (Smith et al., 2013). In particular, some broad key functional gene categories involved in DOM utilization (Poretsky et al., 2010; Rinta-Kanto et al., 2012) and specific functional gene groups linked to successive decomposition of phytoplankton blooms (Teeling et al., 2012) are significantly different, indicating the fundamental differences in survival strategies in relation to potentially available substrates. It is further revealed that PA microbes generally have larger genomes with a variety of metabolic and regulatory capabilities of utilizing compositionally varied organic matter, while the genomes of FL microbes usually are smaller with streamlined metabolic and regulatory functions that enable efficient adaption to oligotrophic conditions (Smith et al., 2013; Yawata et al., 2014; Yung et al., 2016). Phylogenetically, PA and FL lineages generally exhibit different compositions. The PA fraction is relatively enriched in members of Gammaproteobacteria, Verrucomicrobia, Bacteroidetes, Firmicutes and Planctomycetes (Azam and Malfatti, 2007; Milici et al., 2016; Salazar et al., 2016; Suter et al., 2018), while the FL assemblages are often populated by members of Alphaproteobacteria (SAR11 clade or Candidatus Pelagibacter) and Deferribacteres (DeLong et al., 1993; Crespo et al., 2013; Milici et al., 2017). However, significantly overlapped compositions of PA and FL microbial communities were also reported in a few studies (Hollibaugh et al., 2000; Ghiglione et al., 2007; Ortega-Retuerta et al., 2013; Rieck et al., 2015; Liu et al., 2018a). Actually, most members of the PA and FL clades are generalists which switch their lifestyles via attachment and detachment to particles (Crespo et al., 2013; Li et al., 2015). As revealed in many marine niches, Alphaproteobacteria, Gammaproteobacteria and Bacteroidetes are the major overlapped phylum and classes in both PA and FL microbial fractions (Yung et al., 2016).
Our current knowledge of PA and FL microbial populations largely relies on the upper photic ocean, whereas little information is known from the deep dark ocean, which is the largest biome and accommodates more than half of the ocean's microbes (Aristegui et al., 2009; Salazar et al., 2016). Recently, a number of studies have revealed the PA and FL microbial communities in the bathypelagic waters (Li et al., 2015; Salazar et al., 2015; Milici et al., 2017; Mestre et al., 2018) or the deepest abyssal and hadal environments (Eloe et al., 2011; Tarn et al., 2016; Liu et al., 2018a). It is shown that PA and FL bacterial communities in the deep ocean have clear differences in abundance and composition, in addition to the detection of novel, unknown prokaryotic taxa. Furthermore, although archaea are a major component of the marine ecosystem and play significant roles in the degradation of organic materials (Iverson et al., 2012; Suzuki et al., 2017), PA and FL archaeal communities receive less attention and little is known about them. Previous limited reports have observed controversial results, as several studies showed no obvious differences in archaeal community structures between PA and FL assemblages (Galand et al., 2008; Eloe et al., 2011; Suzuki et al., 2017), while a clear separation was found in recent reports (Tarn et al., 2016), with the PA archaeal fraction dominated by Marine Group II (MGII) and Marine Group III (MGIII) and the FL archaeal fraction dominated by Marine Group I (MGI) and anaerobic methane-oxidizing archaea (ANME). In brief, the changes of PA and FL prokaryotes along vertical profiles of the water column, from the surface to the deep bathyal, abyssal and hadal depths, are not well known.
In this study, we analyzed and compared microbial compositions between PA and FL fractions at different depths along the vertical profile in the South China Sea (SCS). The SCS is a marginal sea located in the northwestern Pacific Ocean with a maximal depth of approximately 5380 m (Fig. S1). Our results reveal diverse and significantly divergent microbial compositions in PA and FL fractions and obvious community stratification at different depths along the vertical profiles.
2.1 Sample collection and environmental parameter measurements
Seawater samples were collected from two stations – the G3 station, at a depth of 4039 m at 117∘00.131′ E, 16∘59.947′ N, and the J5 station, at a depth of 4301 m at 114∘00.209′ E, 13∘59.958′ N – located in the central deep basin of the SCS during the open cruise of R/V Dongfanghong II from 3 to 18 July 2014 (Fig. S1). Both stations have a depth > 4000 m, providing us the bathyal environments to vertically profile the variation of microbial assemblages with depth. A Sea-Bird CTD rosette sampler (SBE 911 plus) with 12 L Niskin bottles (Seattle, Washington, USA) was used to collect seawater from six different depths (50, 200, 1000, 2000, 3000 and 4000 m) at each station.
Basic environmental parameters of the water column, including depth, salinity, temperature and dissolved oxygen (DO), were obtained in situ using the conductivity–temperature–depth (CTD) profiler and a DO sensor during the sampling. Once water samples were collected on board, about 0.1 L of seawater was taken immediately for pH measurement using a pH meter (Mettle Toledo Inc., Switzerland).
Approximately 8 L of seawater was filtered on board through a Φ142 mm precombusted glass fiber membrane (0.7 µm nominal pore size, Whatman, USA) under a gentle vacuum of < 150 mm Hg for particulate organic carbon (POC) collection. The membranes were folded and stored at −20 ∘C until our POC analysis. Then about 30 mL of filtered seawater of each sample was collected into 40 mL precombusted EPA vials and stored at −20 ∘C immediately for dissolved organic carbon (DOC) concentration measurement (laboratory on land). About 200 mL filtered seawater at each depth was stored at −20 ∘C for analysis of nutrients (NO ∕ NO, dissolved inorganic phosphate and silicate). The remaining seawater was stored at −20 ∘C for other analyses.
At each depth, we collected 4 L of seawater to obtain microorganisms for further analysis. Seawater was filtered first through a Φ47 mm polycarbonate (PC) membrane of 3.0 µm nominal pore size (Millipore, USA) and subsequently through a Φ47 mm PC membrane of 0.22 µm nominal pore size (Millipore, USA) to collect the PA and FL microorganisms, respectively (Eloe et al., 2011). To avoid damaging the membrane and the fragile particles, a relatively low vacuum pressure of < 10 mm Hg was used, and at the same time, the filtration time was no longer than 40 min for each membrane. The membranes were then frozen at −80 ∘C until further microbial analysis.
Concentration of POC was determined with a PE2400 Series II CHNS/O analyzer (PerkinElmer, USA) (Chen et al., 2008). DOC concentration was measured using a Shimadzu TOC-V analyzer (Shimadzu Inc., Japan) (Meng et al., 2017). Nutrients were determined using a four-channel continuous-flow Technicon AA3 auto-analyzer (Bran+Luebbe GmbH, Germany).
About 1 L of seawater for each sample was sent to Beta Analytic, Inc. in Miami, Florida, for 14C radiocarbon dating with the accelerator mass spectrometry (AMS) method as described in their website (https://www.radiocarbon.com/beta-lab.htm, last access: 2 December 2014). When CTD rosette sampler came back on board, seawater for 14C dating was taken from Niskin bottles with first priority. To avoid the disturbance of air during the sampling, glass bottles were fully filled with flowing seawater with as little head space as possible. In addition, mercury chloride was added to prevent any microbiological influence.
2.2 DNA extraction
In this study, we used the SDS-based method to extract the total DNA as described by Li et al. (2015) with minor modifications. The PC membranes containing seawater microbes were first cut into small pieces in a sterile petri dish and put into autoclaved 2 mL centrifuge tubes. A total of 800 µL of DNA extraction buffer consisting of 100 mM Tris-HCl, 100 mM sodium EDTA, 100 mM sodium phosphate, 1.5 M NaCl and 1 % hexadecyl trimethyl ammonium bromide (CTAB) was added into each tube. The centrifuge tubes were frozen in liquid nitrogen and then thawed in a 65 ∘C water bath. This procedure was repeated three times. When the centrifuge tubes cooled down to room temperature, proteinase K was added with a final concentration of ∼ 0.2 mg mL−1. The tubes were then incubated in a 65 ∘C water bath for 2 h and shaken gently every about 30 min. Then, 800 µL of phenol : chloroform : isoamyl alcohol (, v∕v) was added into the centrifuge tubes, and the tubes were shaken gently several times and centrifuged at 12 000×g for 10 min. The supernatant was carefully transferred into new tubes, and an equal volume of chloroform ∕ isoamyl alcohol (24:1, v∕v) was added. The tubes were centrifuged at 12 000×g for 10 min. The aqueous layer was pipetted into clean 2 mL tubes, and 0.6 volume of cold isopropanol and 0.1 volume of 3 M sodium acetate were added. The centrifuge tubes were incubated at −20 ∘C for 1 h and centrifuged at 12 000×g for 10 min. The liquids were carefully discarded, and DNA pellets at the bottom were gently rinsed with 70 % pre-cooling ethanol. Finally, each DNA pellet was suspended into sterile deionized H2O with a volume of 50 µL.
2.3 Pyrosequencing and analysis of 16S rRNA gene sequence amplicons
Before PCR amplification, we first used the PicoGreen dsDNA Quantitation Kit (Life Technologies, USA) to quantify the concentration of DNA. DNA concentrations obtained varied between 4.48 and 29.1 ng µL−1 with a volume of ∼ 50 µL for each sample. For the PCR amplification of the bacterial 16S rRNA gene, the primer sets 27F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and 533R (5′-TTA CCG CGG CTG CTG GCA C-3′) with 10-nucleotide barcodes were used, while Arch344F (5′-ACG GGG YGC AGC AGG CGC GA-3′) and Arch915R (5′-GTG CTC CCC CGC CAA TTC CT-3′) containing 8-nucleotide barcodes were used for archaea (Ohene-Adjei et al., 2007; Sun et al., 2014). About 10 ng of DNA template was amplified for PCR. The PCR conditions are as follows: firstly, 94 ∘C, 5 min; then, 94 ∘C, 5 s, 53 ∘C, 50 s, and 72 ∘C, 50 s, total 25 cycles; 72 ∘C, 6 min. The products after PCR amplification were purified with the MiniBEST DNA Fragment Purification Kit (Takara Bio Inc, Japan) and then quantified using the NanoDrop 2000 (Thermo Scientific, USA). The pyrosequencing was carried out at the Majorbio Bio-Pharm Technology, Co., Ltd. (Shanghai, China) with the 454 GS-FLX Titanium system (Roche, Switzerland).
QIIME 1.9.1 was used to perform the following phylogenetic analysis of pyrosequenced amplicons (Caporaso et al., 2010). As described in our previous study (Li et al., 2017), the low-quality reads were first filtered with the following quantity control (QC) criteria: (1) the reads with ambiguous nucleotides, (2) the length of reads < 200 bp, (3) the reads containing > 5 bp homopolymers, and (4) the reads with an average flowgram score < 25 in a quality window of 50 bp. The operational taxonomic units (OTUs) were generated based on 3 % cutoff of sequence similarity, and the longest sequence was picked as the representative sequence of each OTU for downstream analysis. The RDP classifier was used for the taxonomy assignment by the SILVA 16S rRNA gene database (version 132). The ChimeraSlayer in the QIIME package was used to identify and exclude those of potential chimeras after alignment with PyNAST. In addition, the singletons were removed from the final OTU tables.
2.4 Diversity estimators and statistical analyses of microbial communities
To avoid the variation caused by an unequal sequence number across samples, the OTU abundance was normalized by resampling of sequences for each sample based on the sample with the fewest number of sequences. After resampling the sequences to the same number, diversity estimators including Chao 1 and Shannon's diversity (H) indices were calculated. Similarities among different microbial communities were determined using similarity matrices generated according to the phylogenetic distance between reads (UniFrac distance), and beta diversity of principal coordinates analysis (PCoA) was computed as components of the QIIME pipeline. The correlation between the microbial community structures and environmental parameters was analyzed by canonical correspondence analysis (CCA). For the PCoA and CCA ordinations, the transformation of the resampled OTU abundance table was performed by taking the log of the sequence numbers. In addition, to test the statistical significance of different groups identified by PCoA ordination, multiple statistical analyses including MRPP, ANOSIM and PERMANOVA were performed based on the resampled and transformed OTU abundance table. A Mantel test was also performed to test the statistical significance of environmental factors with microbial community compositions from the results of CCA. All statistical analyses were performed in the R environment (v 3.2.1) using the vegan package (https://CRAN.R-project.org/package=vegan, last access: 4 February 2019).
In this study, we used the “odds ratio” to assess microbial preference for the PA or FL lifestyles. As defined by Ganesh et al. (2014), the formula of the odds ratio is the following:
where a positive value indicates the PA preference, while a negative value signifies the FL preference (Suter et al., 2018).
2.5 Quantification of 16S rRNA gene and cell abundance estimation
The copy numbers of the microbial 16S rRNA gene for PA and FL fractions were estimated with the 7500 Real-Time PCR System (Applied Biosystems, Thermo Fisher, UK). The primer sets used were 341f and 518r for bacteria (Dilly et al., 2004) and 344f and 519r for archaea (Bano et al., 2004) with about 200 bp amplified DNA fragments. The PCR products of the bacterial and archaeal 16S rRNA gene were first cloned into a pUC18 plasmid vector (Takara Bio Inc, Japan) and then transformed into E. coli DH5α. The recombinant plasmids were extracted and purified and subsequently diluted 10-fold as the standard for real-time PCR. R2 for the standard curves varied between 0.994 and 0.996, indicating a linear relationship well over the concentration ranges used in our study. PCR was carried out in a 20 µL amplification volume. The reaction mixture contained 1 µL of DNA template, 0.15 µM forward and reverse primers, and 10 µL of Power SYBR Green PCR Master Mix (Life Technologies, USA). The PCR amplification conditions include the following: 95 ∘C, 10 min to activate polymerase; 95 ∘C, 15 s, 60 ∘C, 1 min, 40 cycles. A negative control was used to monitor potential contamination, and agarose gel electrophoresis helped to confirm the absence of nonspecific amplification. Melt curves were generated using the Applied Biosystems real-time PCR system software with default thresholds. Each sample had triplicate amplifications, and the average copy number of the 16S rRNA gene was calculated. To make a direct comparison between bacterial and archaeal abundances, we converted the copy number of the 16S rRNA gene into cell abundance based on the assumption that, on average, a bacterial cell has 4.08 16S rRNA gene copies while archaea contain 1.71 copies per cell (Lee et al., 2009). Although the cell abundances inferred from the 16S rRNA gene copy number quantified by qPCR may be potentially biased, the estimation of cell abundances based on the qPCR of the 16S rRNA gene has been confirmed as an effective method to reflect the approximate cell abundances in previous studies.
3.1 Environmental parameters of the water columns
Fundamental environmental parameters, including temperature, salinity, pH, DO, DOC and POC, are listed in Table 1. In general, they showed similar vertical trends with the normal pelagic ocean. Salinity increased gradually from ∼ 33.84 at 50 m to ∼ 34.52 at 200 and 1000 m and then remained at around 34.60 at greater depths until 4000 m. DO concentration was the highest (∼ 204.5 µM) in surface water, decreased gradually to the lowest (∼ 83.9 µM) at 1000 m depth and then increased gradually from ∼ 102.0 µM at 2000 m to ∼ 113.5 µM at 4000 m. Nitrite concentrations of the water columns at all depths were below the detection limit. Concentrations of nitrate, phosphate, and silicate were continuously increasing from the surface to 1000 m depth and then remained at relatively constant levels (Table 1).
Table 1Environmental parameters of the water columns at different depths of G3 and J5 stations in the South China Sea.
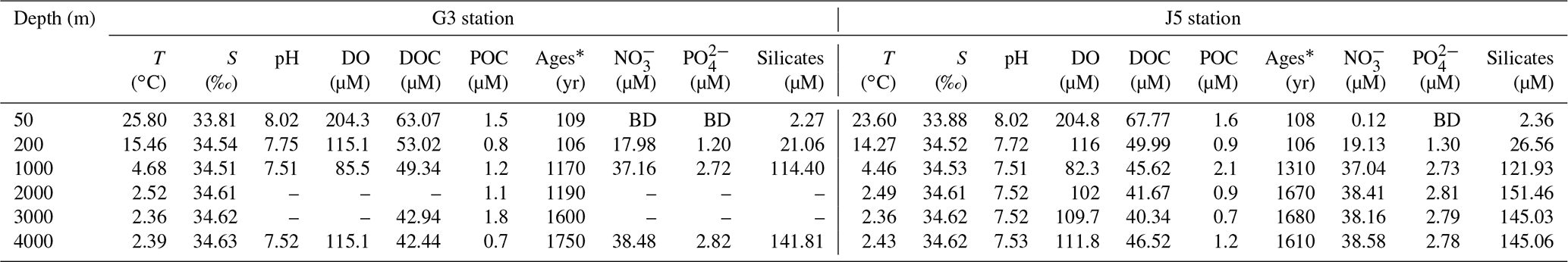
* 14C ages; BD: below detection; –: no measurement.
As expected, the age of the seawater determined from Δ14CDIC was youngest at the surface and increased with depth linearly, varying from about 106 to 1650 years. The upper water layers (50 and 200 m) from the two stations had the youngest and nearly the same ages, around 106 years. Ages of 1000 and 2000 m in the G3 station were almost identical, around 1180 years, and increased to 1600 years at 3000 m and 1750 years at 4000 m. By contrast, the age of 1000 m in the J5 station was ∼ 1310 years and remained relatively stable below 1000 m with the age of about 1650 years (Table 1). DOC concentrations ranged from 63.07 to 40.34 µmol L−1, with the highest at the surface and the lowest at the deep. However, POC concentrations varied greatly between 0.5 and 2.1 µmol L−1 and showed great variations. The POC concentrations were highest at 3000 m of the G3 station (1.8 µmol L−1) and at 1000 m of the J5 station (2.1 µmol L−1) (Table 1).
3.2 Microbial cell abundances
The estimated abundances of bacteria and archaea were about 106–109 cells L−1 and 105–107 cells L−1, respectively (Fig. 1). The FL bacterial fraction generally accommodated higher cell abundances (varying from 0.62 × 107 to 1.65 × 108 cells L−1), several times higher than their corresponding PA fraction (1.85 ± 0.02 × 106–1.90 × 108 cells L−1). However, one remarkably lower abundance of the FL bacterial fraction than the PA fraction was detected in the surface water (50 m) of the G3 station, where PA bacterial abundance was up to 1.70 × 109 cells L−1, 2 orders of magnitude higher than that of the FL fraction (1.62 × 107 cells L−1) (Fig. 1a). Similar to bacteria, the FL archaeal fractions usually showed higher cell abundances than their PA fractions (Fig. 1b). The only exception was also at the depth of 50 m of the G3 station, where the estimated PA archaeal cell abundance (6.50 ± 0.01 × 107 cells L−1) was much higher than that of the FL archaeal fraction (1.01 × 106 cells L−1). The FL archaeal fraction had cell abundances between 2.70 × 105 and 8.62 ± 0.03 × 106 cells L−1, while PA archaeal fractions fluctuated between 1.28 × 105 and 6.50 ± 0.01 × 107 cells L−1 (Fig. 1).
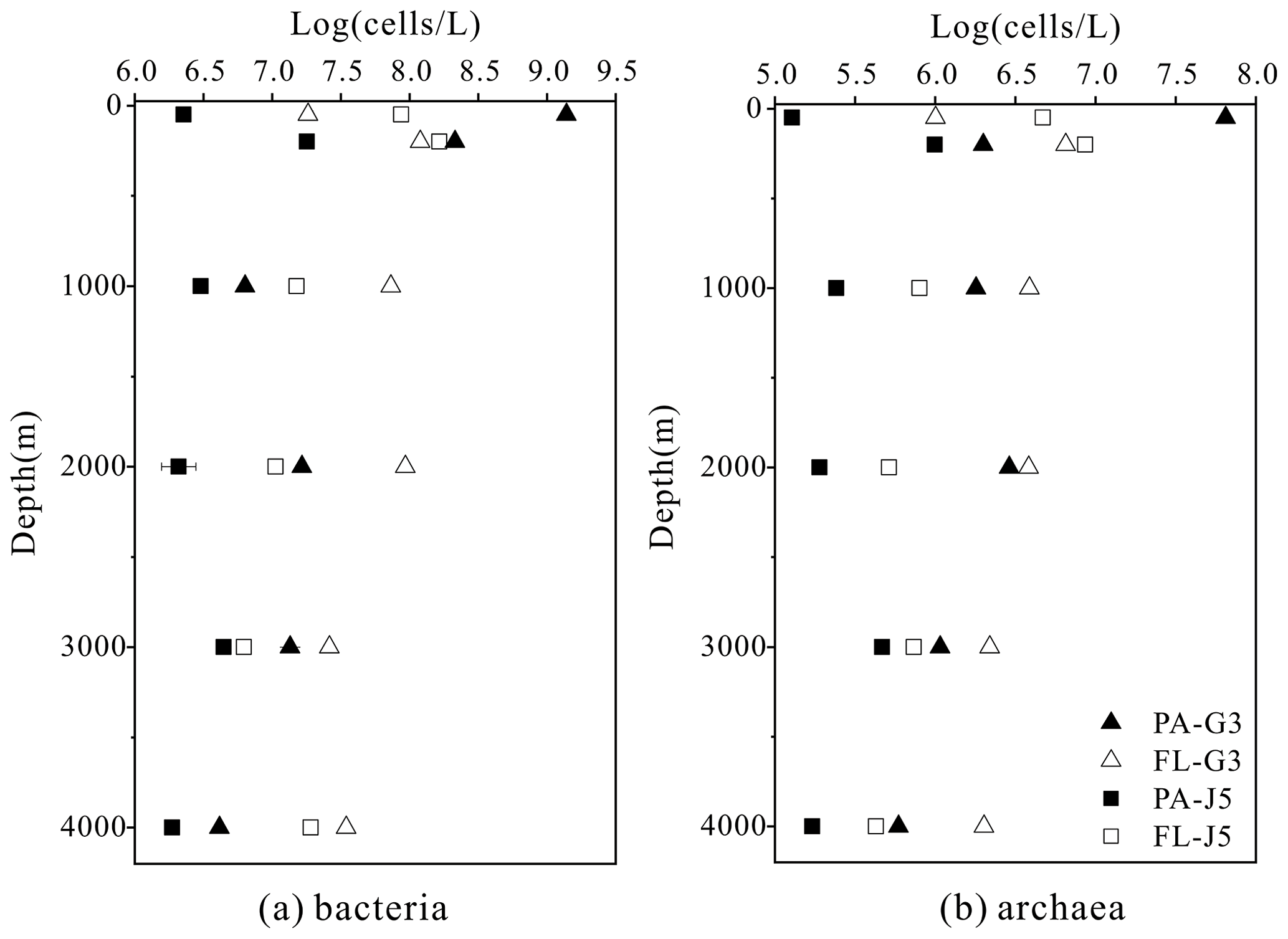
Figure 1Bacterial and archaeal cell abundances in seawaters at different depths from the G3 station and J5 station in the South China Sea, estimated from 16S rRNA gene copy abundances.
3.3 Estimation of microbial diversity
In total, (91 692, 81 332) and (72 590, 93 059) valid sequences of the bacterial 16S rRNA gene were obtained for FL and PA fractions of G3 and J5 stations, respectively. Based on the 97 % similarity, these FL and PA bacterial sequences were defined into a total of 6320 operational taxonomic units (OTUs) in which 1982 OTUs belonged to singletons and were finally removed from the valid OTU table (Table S1). Correspondingly, (50 727, 41 511) and (44 443, 37 751) archaeal sequences were determined for FL and PA archaeal fractions of the G3 and J5 stations. Attempts to determine the PA archaeal sequence from 3000 m depth of the G3 station and 4000 m depth of the J5 station failed because of technical reasons. A total of 1070 archaeal OTUs were defined, and 329 OTUs were considered as singletons (Table S2). The sequencing depths of the 16S rRNA gene were shown in their rarefaction curves (Fig. S2).
Shannon's diversity (H) and Chao 1 indices were calculated to estimate microbial diversity of both PA and FL fractions at all depths (Figs. 2 and S3). In most cases, the H indices of the bacterial FL fractions were usually higher than their PA counterparts at each depth (Fig. 2). The H index of FL and PA bacterial fractions gradually increased from 50 to 1000 m, decreased in the intermediate water of around 2000 m depth, and increased again at 3000 and 4000 m (Fig. 2a). The archaeal H index varied along the vertical profiles with a trend similar to bacteria, and FL archaea generally had higher H index values than the PA fraction (Fig. 2b). In addition, it was further observed that even at the same depth the values of the H index between two stations fluctuated a lot. The Chao 1 index showed nearly similar variation trends for both PA and FL microbial fractions (Fig. S3).
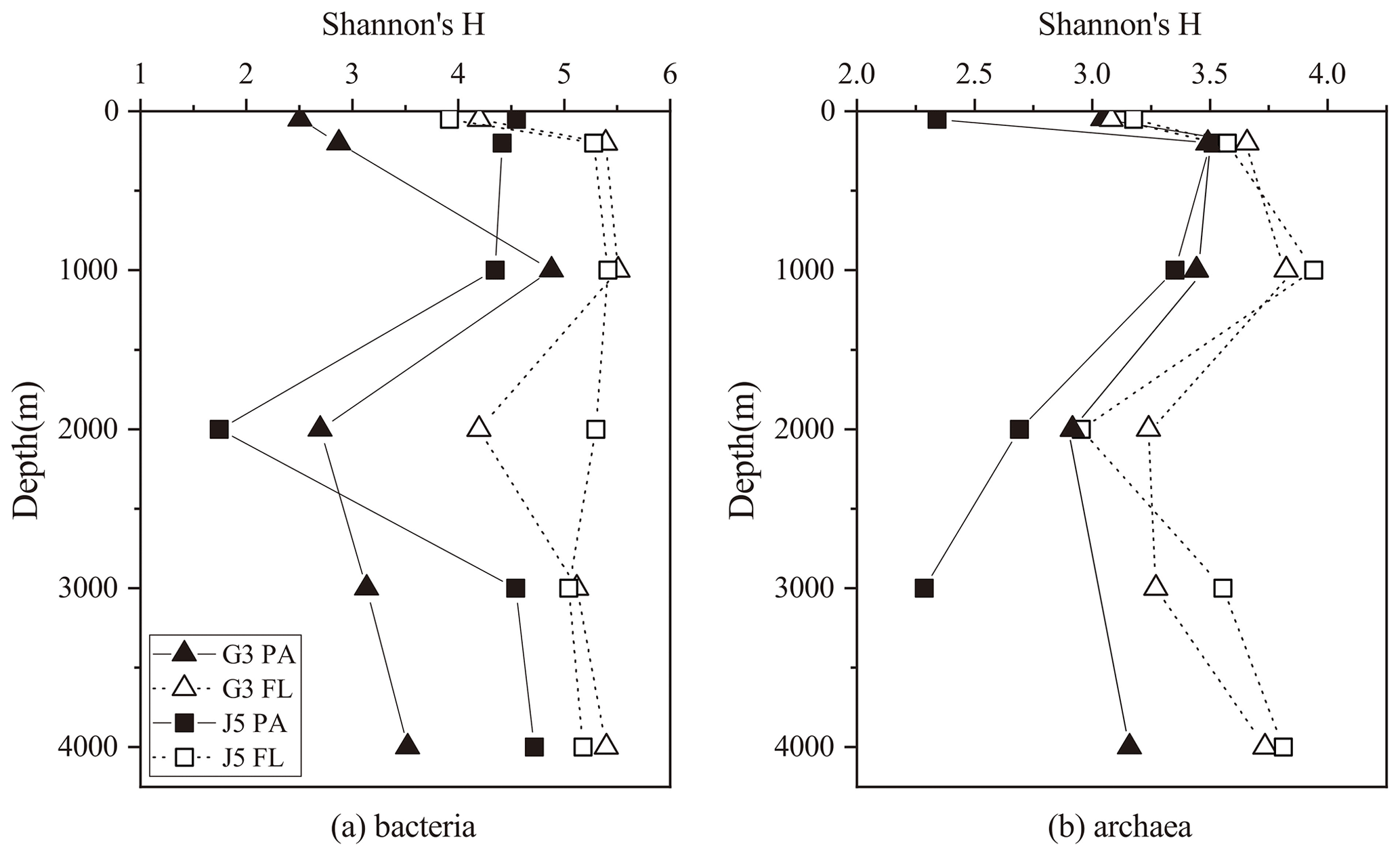
Figure 2Shannon's diversity index calculated for all bacterial and archaeal communities of seawaters collected from the G3 station and J5 station in the South China Sea.
PCoA analysis revealed that there were significant differences (P values < 0.05, Table S3) in bacteria and archaea community structures over the depth profiles and between the FL and PA fractions. Overall, three groups were distinguished, the surficial 50 m group, the FL group, and the PA group (Fig. 3). One group, which consisted exclusively of samples at 50 m depth, separated the microbes at the surface from those in the rest of the water column of both stations, irrespective of microbial lifestyles (FL or PA). However, the other two groups were separated mainly based on the FL and PA lifestyles. It is interesting to note that the FL bacterial samples clustered into one group where samples were further partitioned with respect to depth (Fig. 3a). Canonical correspondence analysis (CCA) showed that fundamental environmental parameters including depth, DO, salinity, seawater age, DOC and POC concentration, and silicate exerted potential impact on variations of FL and PA microbial communities along the water column (Figs. 4, S4). A Mantel test further indicated that all those factors, except POC concentration (P=0.164), were the statistically significant variables associated with variation of PA and FL fractions (P=0.001).
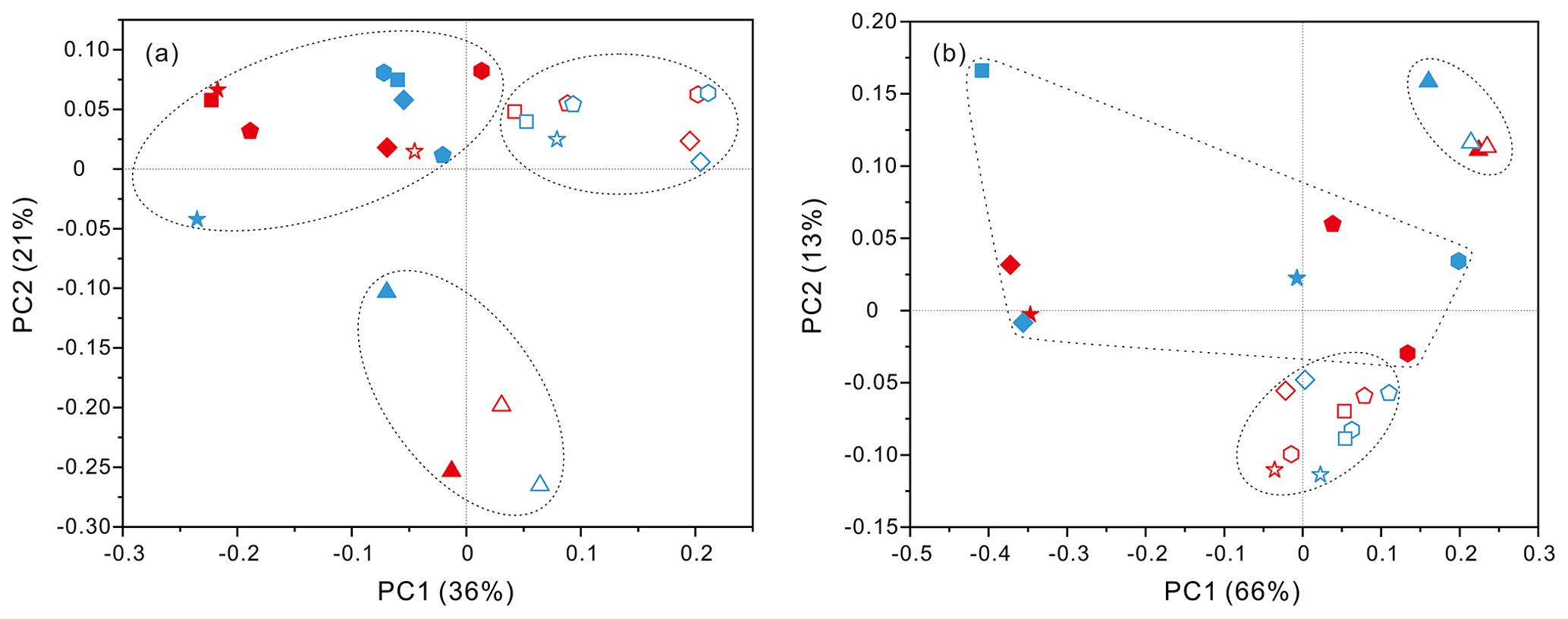
Figure 3Results of PCoA analysis for particle-attached and free-living microbial fractions collected from seawater columns of the South China Sea. (a) PA and FL bacteria; (b) PA and FL archaea. Statistical analyses supported the groups with statistical significances (Table S3). Triangle: 50 m; rhombus: 200 m; hexagon: 1000 m; star: 2000 m; square: 3000 m; pentagon: 4000 m. Blue color: J5 station; red color: G3 station. Filled: particle-attached fraction; open: free-living fraction.

Figure 4Results of CCA analysis to correlate several environmental factors including POC, seawater age, salinity and depth to PA and FL microbial communities collected from seawater columns of the South China Sea. (a) PA and FL bacteria; (b) PA and FL archaea. Triangle: 50 m; rhombus: 200 m; hexagon: 1000 m; star: 2000 m; square: 3000 m; pentagon: 4000 m. Blue color: J5 station; red color: G3 station. Filled: particle-attached fraction; open: free-living fraction.
3.4 Taxonomic compositions of the PA and FL bacterial and archaeal fractions
Taxonomic compositions of FL and PA bacterial fractions and their relative abundances are presented in Fig. 5. At the phylum level, bacterial sequences were mainly assigned into Proteobacteria (Alpha-, Beta-, Gamma- and Deltaproteobacteria), Actinobacteria, Cyanobacteria, Planctomycetes, Bacteroidetes, Marinimicrobia (SAR406 clade), Chloroflexi, Firmicutes, Acidobacteria, Gemmatimonadetes, Nitrospinae and Verrucomicrobia. The taxa at the family level with relatively high abundances (> 3 %) on average in either the PA or FL fraction are further shown in Fig. 6.
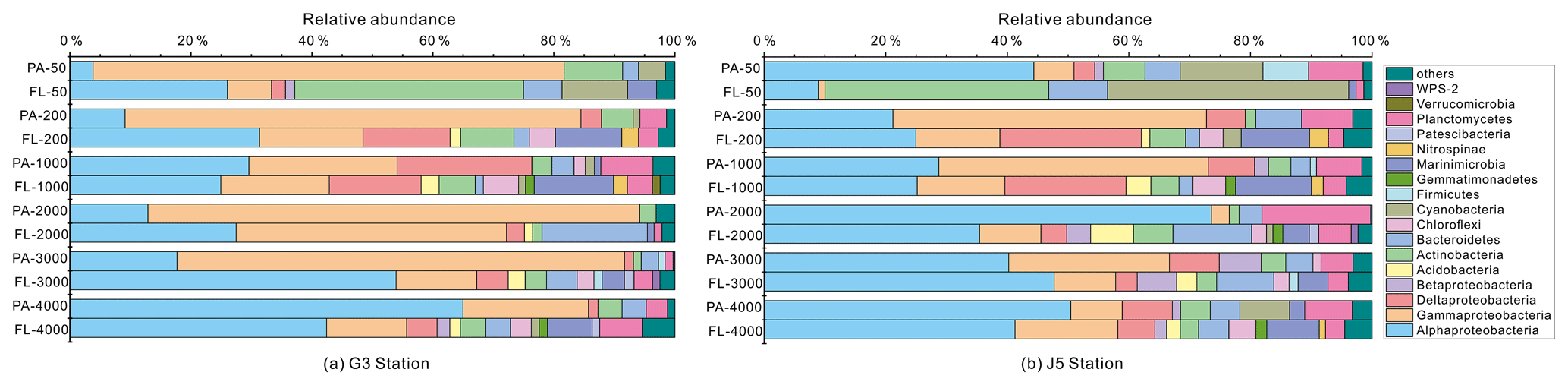
Figure 5Taxonomic compositions of particle-attached and free-living bacterial communities of seawaters at different depths along two different water columns in the South China Sea. (a) G3 station; (b) J5 station. The phylum or class which has less than 1 % proportions is classified into “others” (Fig. S5).
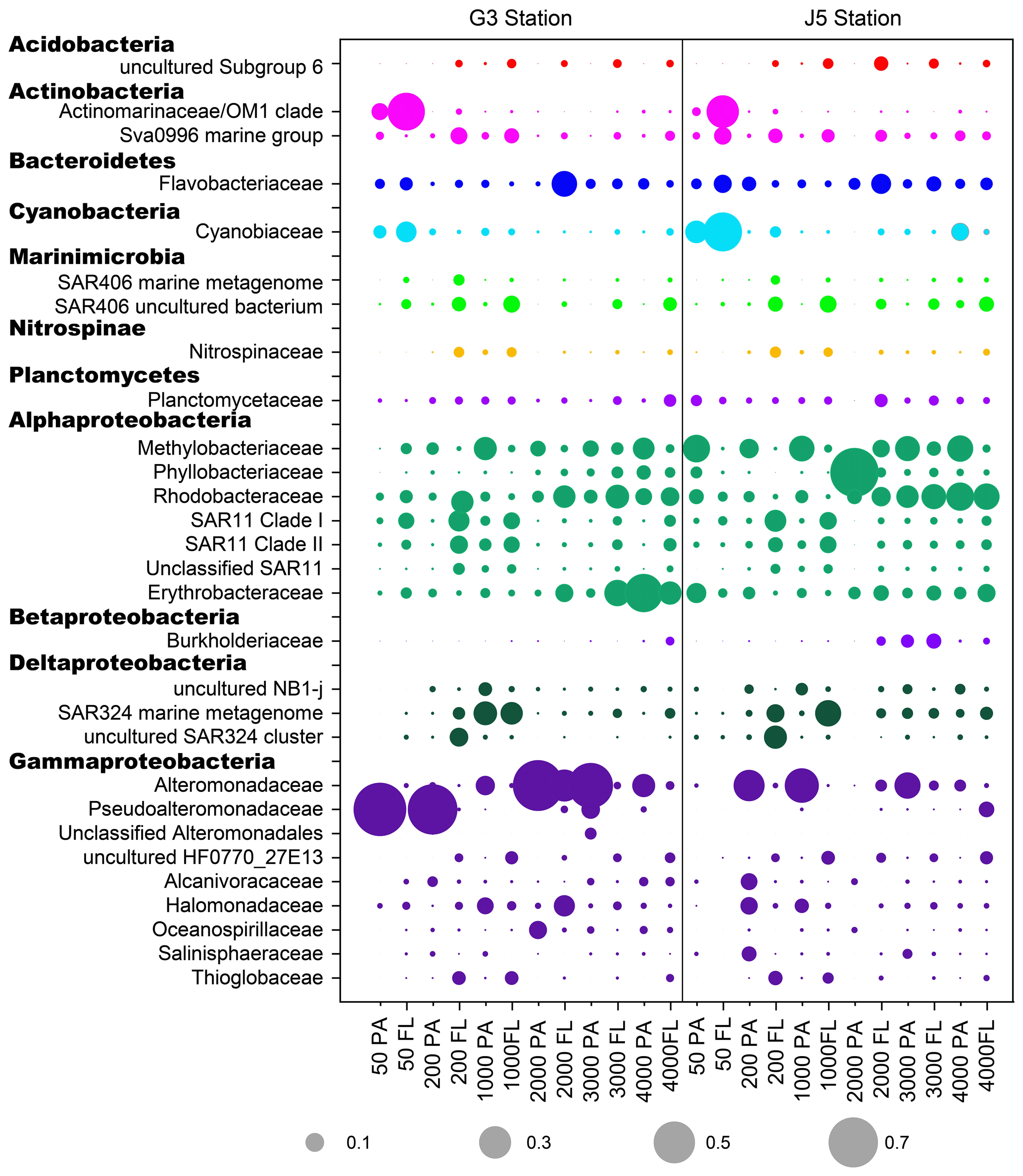
Figure 6The relative abundances of families in particle-attached and free-living bacterial communities. Dark grey bubbles are the average relative abundances in the PA fraction, while light grey bubbles are the average relative abundances in the FL fractions. Scale is shown at the bottom, and the circle with a number indicates the actual relative abundance.
It is clear that Alphaproteobacteria and Gammaproteobacteria were the dominant lineages in both the FL and PA fractions at nearly all depths. In most cases, the sum of Alphaproteobacteria and Gammaproteobacteria accounted for ∼ 40 % to nearly 90 % (Fig. 5). Moreover, their relative abundances in different PA and FL fractions and different stations also varied widely. Within the Alphaproteobacteria, the dominant families included Methylobacteriaceae, Phyllobacteriaceae, Rhodobacteraceae and Erythrobacteraceae (Fig. 6). Members of the families Methylobacteriaceae and Erythrobacteraceae occurred commonly in both fractions at almost all depths but usually with higher proportions in PA fractions. The family Rhodobacteraceae occurred commonly in both fractions at every depth (1 %–20 %), while the Phyllobacteriaceae was dominantly distributed in the PA fraction of 2000 m depth of the J5 station with > 60 % proportions. In addition, another important lineage within Alphaproteobacteria is the SAR11 clade (now named Pelagibacterales) (Grote et al., 2012). It was clearly revealed that the SAR11 clade showed relatively higher abundances in FL fractions than PA fractions. Moreover, at depths above 1000 m, the SAR11 clade had a far higher proportion than the deep ocean, and the maximum levels occurred at 200 m depth (20 %–24 %) (Fig. 6, Table S1). Gammaproteobacteria was another lineage with the highest abundance overall. Its relative abundances changed significantly with depths and in different fractions. The minimum abundances were only 1 %–5 %, while the maximum were up to 73 %–80 % (Fig. 5 and Table S1). Moreover, the G3 station generally had higher Gammaproteobacteria proportions than that of the J5 station on average. As shown in Fig. 6, although sequences of Gammaproteobacteria were classified into multiple families, actually only two families, Alteromonadaceae and Pseudoalteromonadaceae, exhibited absolutely dominant prevalence in the bacterial populations. The Pseudoalteromonadaceae populated predominantly the PA fractions in 50 and 200 m depths (66 %–75 %), while the Alteromonadaceae mainly dominated the PA fractions in the deep water, particularly at 2000 and 3000 m depths. Deltaproteobacteria also had a common distribution in both fractions of all depths, usually accounting for less than 10 % proportions in most samples (Fig. 5), and SAR324 clade members contributed significantly to the dominance of the Deltaproteobacteria (Fig. 6). Actinobacteria and Cyanobacteria were abundantly distributed only in the surficial 50 m depth, and by sharp contrast, their proportions in other depths were less than 5 %. Other bacterial lineages which had a wide distribution in all depths but only with minor abundances in both fractions included Planctomycetes, Bacteroidetes, Marinimicrobia (SAR406 clade), Chloroflexi, Betaproteobacteria, Firmicutes, Gemmatimonadetes and Verrucomicrobia (Fig. S5).
The majority of archaeal amplicons mainly fell into the Nitrosopumilales and several uncultured taxonomic lineages (Figs. 7 and S6). Both FL and PA archaeal fractions at all depths were principally populated by the order Nitrosopumilales (formerly referring to MGI.1a, a subclade of MGI) (Qin et al., 2017) of the Thaumarchaeota and Marine Group II (MGII) of the Euryarchaeota. Members from the Nitrosopumilales and MGII lineages generally contributed more than 80 % relative abundances in their respective clone libraries. The Nitrosopumilales was always one of the most abundant clades along the vertical profiles except in the topmost FL and PA fractions. The MGII clade exhibited a wide distribution along the water columns, and it usually accounted for the large proportions in both archaeal size fractions. The photic layer (∼ 50 m depth) contained the highest abundances of the MGII clade, particularly in FL fractions with up to ∼ 80 % proportions. By sharp contrast, the lowest abundances of MGII occurred at 2000 (G3 station) and 3000 m (J5 station) depths, making up < 20 % of proportions. The third most abundant clade overall is Marine Group III (MGIII) of the Euryarchaeota. MGIII representatives were mainly dispersed in the FL fractions with 5 %–18 % abundances, while they were absent from most of the PA fractions. However, the relative abundances of MGIII members in PA fractions of 1000 m depth could be as high as 30 %–45 % (Fig. 7). The order Methanosarcinales of Euryarchaeota was detected commonly in most PA fractions, but it had the higher abundance only in the upmost 50 m depth (∼ 29.7 %) (Fig. 7). Another sample accommodating relatively many Methanosarcinales was the PA faction of 3000 m in the J5 station with 9.1 % proportion. Within the Euryarchaeota, another clade of methanogens, Methanobacteriales, was also detected from both size fractions but with low relative abundances (< 5 %) (Figs. 7, S6, Table S2). In addition, other archaeal lineages included Woesearchaeota (formerly known as the DHVEG-6 group), the Miscellaneous Crenarchaeotic Group (MCG, now named Bathyarchaeota), the Halobacteriales of the Euryarchaeota and Marine Benthic Group A (MBG-A) of the Thaumarchaeota. They just provided a limited contribution to archaeal populations (Fig. S6).
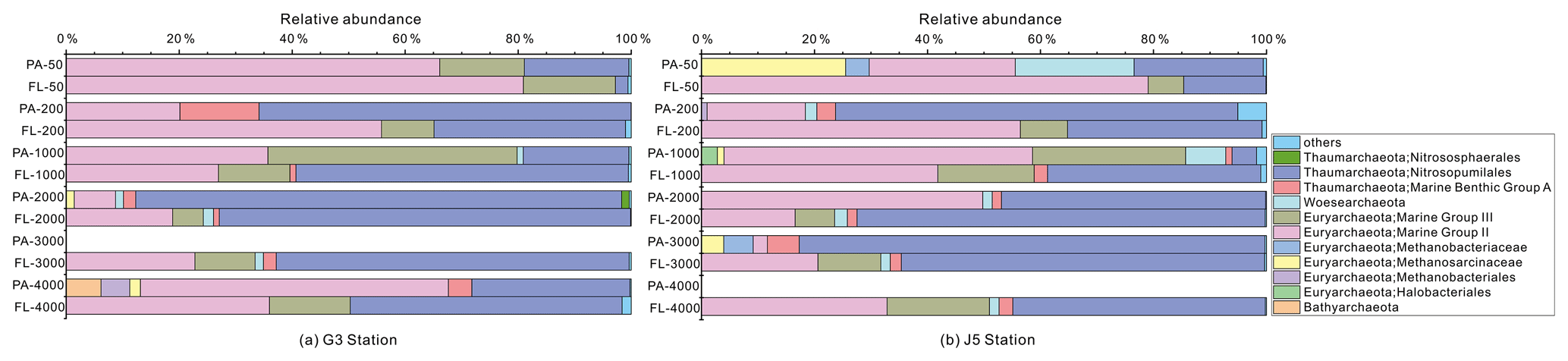
Figure 7Taxonomic compositions of particle-attached and free-living archaeal communities of seawaters at different depths along two different water columns in the South China Sea. (a) G3 station; (b) J5 station. PA-3000 at the G3 station and PA-4000 at the J5 station indicate the samples failing in the sequencing of archaeal 16S rRNA gene. The archaeal lineages, at the phylum or class level, with less than 1 % proportions are classified into “others” (Fig. S6).
3.5 Bacterial preference for PA or FL lifestyles
The odds ratio was used to assess the preference of bacterial taxonomic lineages to the PA or FL lifestyle. A positive odds ratio indicates PA preference or higher abundance in the PA fraction, while a negative value suggests FL preference or higher abundance in the FL fraction. The bacterial lineages dominating the PA fractions come exclusively from Alphaproteobacteria and Gammaproteobacteria with some relatively abundant Deltaproteobacteria and Planctomycetes at specific depths (Fig. 5). By contrast, although the predominant lineages of FL fractions also mainly consisted of members of Alphaproteobacteria and Gammaproteobacteria, other abundant lineages were more diverse, including Actinobacteria, Cyanobacteria, Bacteroidetes, Marinimicrobia and Deltaproteobacteria, as shown in Fig. 5. As shown in Fig. 8, we listed those lineages at the family level with high proportions (> 1 %) with their odds ratios along the depth profiles. It was suggested that most of the absolutely dominant families of PA fractions comprised of the Phyllobacteriaceae and Methylobacteriaceae (Alphaproteobacteria) and Pseudoalteromonadaceae and Alteromonadaceae (Gammaproteobacteria) (Fig. 6) showed a preference for the PA lifestyle. However, the alphaproteobacterial Rhodobacteraceae and Erythrobacteraceae prevailing in PA fractions preferred to different lifestyles at different depths (Fig. 8). Compared with those PA-preferred lineages, there is a wider range of lineages showing a preference for the FL lifestyle. These phylogenetic lineages are mainly populated by the OM1 clade and Sva0996 marine group (Actinobacteria), Nitrospinaceae (Nitrospinae), Planctomycetaceae (Planctomycetes), the SAR11 clade (Alphaproteobacteria), the SAR324 clade (Deltaproteobacteria), the SAR86 clade and Thioglobaceae (Gammaproteobacteria). It is important to point out that a considerable number of bacterial lineages exhibited their preferences for both PA and FL lifestyles, though their preferences varied at different depths or locations (Fig. 8). Actually, at OTU level, less than 1∕2 of the total OTUs (2005 out of 4338 OTUs) were shared by PA and FL fractions (Fig. S7). Phylogenetically, these PA- and FL-shared OTUs mostly fell into Alphaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, Planctomycetes, Chloroflexi, Bacteroidetes, Marinimicrobia and Actinobacteria. The taxonomic components of the PA- and FL-shared OTUs at different levels are approximately similar to OTUs retrieved exclusively from either the PA fractions or the FL fractions (Table S1, Fig. S7).
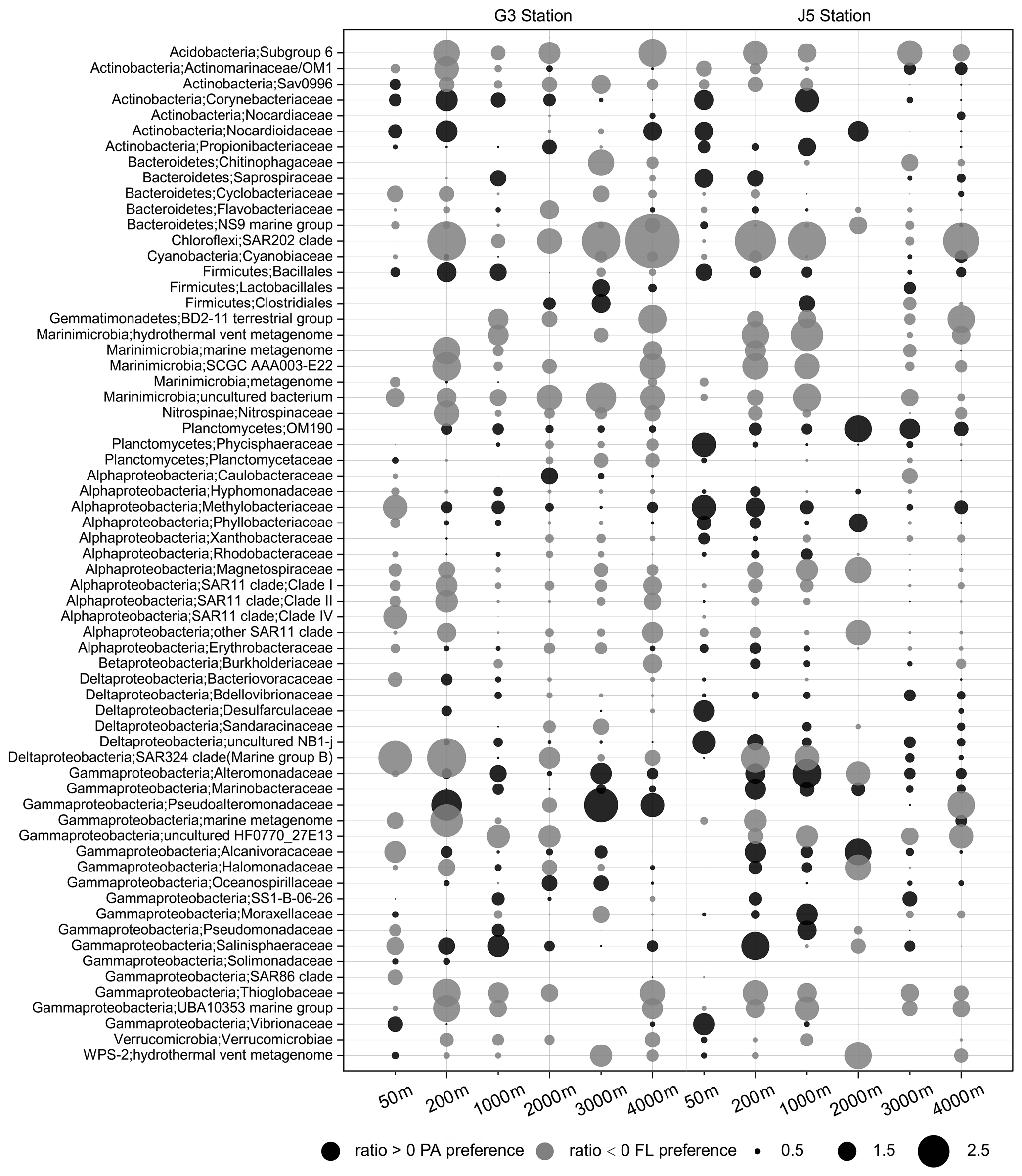
Figure 8Odds ratio for each of the families with relatively abundant proportions in each sample. Dark grey bubbles represent the clades with a positive odds ratio, meaning the preference of the PA lifestyle. Light grey bubbles represent the clades with a negative odds ratio, which is indicative of the FL preference. Scale is shown at the bottom.
4.1 Comparison of microbial abundance and diversity between PA and FL fractions
PA bacterial and archaeal fractions show generally lower abundance and taxonomic richness than their FL counterparts and constitute a small fraction of the total abundances. Our results are consistent in principle with previous reports on various pelagic environments, in either the euphotic zone, twilight or the dark deep ocean (Turley and Stutt, 2000; Simon et al., 2002; Ghiglione et al., 2007; Rieck et al., 2015). However, in some eutrophic and notably particle-rich marine ecosystems, for example, marine snow or estuaries, PA bacterial fractions were present in higher local concentrations and greater diversity than FL bacteria (Caron et al., 1982; Karner and Herndl, 1992; Turley and Mackie, 1994; Garneau et al., 2009). In the upper photic zone, PA bacterial abundance and their contribution to total bacterial biomass are highly variable and depend largely on the quantity and quality of suspended organic particles (Cammen and Walker, 1982; Simon et al., 2002; Doxaran et al., 2012). This is indeed the case in the South China Sea. As shown in Fig. 1, at 50 and 200 m depths of the G3 station, PA bacterial abundances outnumbered FL bacteria by nearly 2–100 times, whereas the J5 station has an opposite trend. However, as shown in Table 1, these two stations have almost the same environmental parameters, particularly in POC concentrations. One possibility may be that G3 and J5 have different POC compositions, attributable to different origins of organic matter (Chen et al., 2015; He et al., 2016; Liang et al., 2018). Although bacteria attaching to particles are of relatively lower abundance compared to free-living cells in the pelagic ocean, they are consistently metabolically more active with higher extracellular enzymatic activities (Karner and Herndl, 1992) and cell-specific thymidine incorporation rates (Turley and Mackie, 1994; Turly and Stutt, 2000). Therefore, PA bacteria often play a comparable role to free-living bacteria in hydrolysis or decomposition of marine organic matter, biomass production, and carbon cycling (Griffith et al., 1994; Turly and Stutt, 2000; Liu et al., 2015). The decline of bacterial abundance and richness along the depth profile is largely owing to the gradual decreasing availability of usable organic carbon (Smith, 1992; Turly and Stutt, 2000; Jiao et al., 2014). It is interesting that the mid-water around 2000 m depth showed the lowest bacterial diversity (Figs. 2, S3). One possibility is that 1500–2000 m is roughly a boundary for different water masses in the deep, central basin of the South China Sea. The deep water masses (> 2600 m) of the central basin coming from the western Pacific Ocean through the Bashi Channel are relatively rich in nutrients compared to the mid-water masses of the SCS. Therefore, it may cause a relative increase in microbial diversity in deep water masses such as those at 3000 and 4000 m. In addition, some “old, deep” water from the bottom of the central basin will also rise to around 2000 m depth because of the basin-scale circulation. These old waters are relatively enriched in refractory DOC (RDOC) and remained after microbial utilization of labile DOC during their circulation, potentially reducing microbial diversity. This hypothesis is partly supported by the seawater age at the J5 station. It is shown that the age of seawater at 2000 m depth of the J5 station is 1670 years, which is roughly equal to those of deep waters at 3000 and 4000 m (1680 years and 1610 year). In contrast, archaea are commonly much lower in cell abundance and community diversity compared with their bacterial counterparts at the same depths (Figs. 1, 2 and S3). The relative abundance of archaeal populations in total prokaryotes increases gradually with depth, which is indicative of a potential rising impact on the biogeochemical cycle in marine environments. In addition, a pronounced distinction in microbial community structures of PA and FL assemblages was observed along the depth profile, which was well supported by the results of statistical analyses (Fig. 3). It is expected that the PA microbial fraction differs from the FL fraction, considering their discrepant activity patterns for survival. Related discussions are shown below.
4.2 Environmental factors potentially shaping microbial community structure
Several environmental parameters played a pivotal role in structuring microbial communities of seawater. Hydrological condition (e.g., depth), together with age and salinity of water mass, is a key subset of environmental drivers (Fig. 4). Recent studies have shown that microbial populations in the meso-/bathypelagic ocean are largely dissimilar to those of the epipelagic zone (Salazar et al., 2015; Milici et al., 2017; Liu et al., 2018a), which is indicative of a crucial environmental selection process exerted by depth. In our study, PCoA analysis revealed that PA and FL fractions from the surficial zone (50 m) were clustered into a separate but relatively loose group distant from other depths (Fig. 3), which is indicative of the influence imposed from depth in shaping microbial community structures. Several bacterial lineages, including Cyanobacteria, Actinobacteria, Deltaproteobacteria, Marinimicrobia (SAR406 clade) and Firmicutes, with distinct distributing stratification contribute to this dissimilarity (Fig. 5). Cyanobacteria and Actinobacteria belong to typical phototrophs (Mizuno et al., 2015), and they are prevalently distributed in euphotic zones. By contrast, the deltaproteobacterial SAR324 clade, as shown in our results, is primarily found in mesopelagic waters (200–1000 m) (Fuhrman and Davis, 1997; Wright et al., 1997). The SAR406 clade has a ubiquitous distribution across diverse marine niches; however, its high abundance always occurs within the mesopelagic zones, ∼ 5 times higher or more than in surface ocean (Yilmaz et al., 2016). Archaeal population components also reflect the impact of depth. Euphotic zones hold less abundant thaumarchaeotal Nitrosopumilales and more euryarchaeotal Methanosarcinales and Woesearchaeota (Fig. 7), while marine thaumarchaeotal groups are more abundant in meso- and bathypelagic waters (Karner et al., 2001; Mincer et al., 2007; Varela et al., 2008). In addition, Salazar et al. (2016) found that sampling depth appears to have a more direct impact on free-living bacterial communities. Our results are highly consistent with this observation in that FL bacterial fractions from the same depth grouped together irrespective of their sampling locations (G3 or J5 station) (Fig. 3a).
DO concentration is observed to strongly affect particle flux and particle transfer efficiency from the euphotic zone to the deep sea since remineralization of organic particles appears to be oxygen dependent (Laufkotter et al., 2017; Cram et al., 2018). DO is considered one of the most crucial environmental variables for shaping the compositions of particle-attached bacterial assemblages (Salazar et al., 2016). Some taxonomic lineages are directly affected by oxygen. For example, a recent study found that oxygen is one of the key factors driving the distribution and evolutionary diversity of Woesearchaeota (Liu et al., 2018b). POC and DOC can be substrates for both PA and FL communities, respectively (Azam and Malfatti, 2007; Zhang et al., 2016; Liu et al., 2018a). However, POC concentration in the present study is not statistically significantly correlated with either bacterial or archaeal community abundances (P values > 0.05) (Table S3). We hypothesize that the quality rather than the quantity of POC imposes a decisive influence on microbial populations, especially in the deep, dark ocean. During POC sinking from surface through the water column, and also as seawater ages, the labile organic matter becomes increasingly decomposed, while the more refractory material remains and resists further degradation (Simon et al., 2002). In such cases, utilization of the POC in the deep sea by microorganisms depends on the quality and quantity of the remaining POC. Meanwhile, in older seawater, DOC also becomes more refractory because free-living microorganisms preferentially utilize labile DOC and the remaining refractory DOC gradually accumulates, which potentially affects microbial community structures. Among common nutrients, silicate exhibited statistically significant correlation with microbial distributions (Fig. S4), and this is unexpected because the SCS generally exhibits N- or P-limited phytoplankton production (Wu et al., 2003; Chen et al., 2004). However, recent research found that, near the sampling site of this study, there is a clear silicon deficiency in the euphotic zones shallower than 75 m (Huang et al., 2015), which directly influences the diversity and biomass of phytoplankton (for example, diatom), and consequently the quantity and quality of POM transported to the deep along the vertical water columns, and finally exerts a potential impact on microbial communities. Some bacterial lineages such as the Rhodobacteraceae, Flavobacteriaceae, Oceanospirillaceae and the SAR11 clade, commonly retrieved in our present study, have been confirmed to be closely related to marine diatom blooms (Zhang et al., 2018; Mönnich et al., 2020). Actually, the microbial community structure and its distribution along the water column profile are a comprehensive combination impacted by multiple environmental variables.
4.3 Specialist or generalist for the PA and FL lifestyle: clues from bacterial community compositions
It was indicated that PA and FL bacterial fractions generally accommodated different community compositions along the depth profiles (Fig. 3), consistent with previous reports in various marine habitats (Acinas et al., 1997; Moeseneder et al., 2001; Ghiglione et al., 2009; Salazar et al., 2015). However, in most cases, the taxonomic compositional disparity between the two filtration fractions does not seem very apparent, at least at the phylum level (Fig. 5). Actually, a few studies also confirmed that at high taxonomic ranks, bacteria show conserved lifestyles either in association with particles or as free-living microorganisms (Eloe et al., 2011; Salazar et al., 2015; Liu et al., 2018a). The pronounced contrast in population compositions of the two filtration fractions was unveiled only at a greater taxonomic level, and a considerable number of phylogenetic taxa exhibited different preferences for PA or FL lifestyles. It is shown in Figs. 5 and 6 that as the most abundant members, Alphaproteobacteria and Gammaproteobacteria, occurred prevalently in both filtration fractions, but at the family level, most of predominant bacterial lineages of PA and FL fractions were significantly divergent, indicating their preference for different microhabitats shaped by organic particles and environmental parameters. The dominant lineages in PA fractions were mainly associated with the families Pseudoalteromonadaceae and Alteromonadaceae within Gammaproteobacteria, as well as the Methylobacteriaceae and Phyllobacteriaceae within Alphaproteobacteria. These gammaproteobacterial members are usually retrieved from diverse marine habitats such as the typical PA clades, and they are believed to have the abilities to degrade/utilize HMW organic compounds with higher nutrient requirements (DeLong et al., 1993; Crespo et al., 2013). The adhesion to particles could make them increase nutrient acquisition and avoid the nutrient-depleted conditions (Crespo et al., 2013). By contrast, members of Alphaproteobacteria are rarely reported as the dominant lineages of the PA fraction or particle-attached preference (Crespo et al., 2013; Rieck et al., 2015; Suzuki et al., 2017), which is inconsistent with our results revealing alphaproteobacterial lineages frequently prevailed as PA members. Further phylogenetic analysis revealed that the majority of alphaproteobacterial PA members belonged to the genus Methylobacterium and are strictly aerobic, facultatively methylotrophic bacteria, which can grow on a wide range of carbon compounds (Green, 2006). They probably benefit from the particle-attached lifestyle, making their high requirements for organic matter easily to achieve. Compared with bacterial PA counterparts, FL bacterial communities are more diverse, and dominant populations are scattered in more phylogenetic taxa with relatively homogeneous proportions (Fig. 8). Among the predominant lineages, the actinobacterial OM1 cade and cyanobacteria dominate the upper surficial waters (Fig. 6), likely attributed to their phototrophic behaviors. Although Actinobacteria are recognized as ubiquitous members of marine bacterioplankton (Giovannoni and Stingl, 2005), they are scarcely reported with predominance (Milici et al., 2016). Ghai et al. (2013) revealed the OM1 clade members possess the smallest cell sizes with streamlined genome, representing a typical adaption to oligotrophic conditions (Giovannoni et al., 2014), which agrees well with the oligotrophic environments in the SCS (Gong et al., 1992). Other predominant FL lineages include the alphaproteobacterial SAR11 clade, the deltaproteobacterial SAR324 clade, and Marinimicrobia (SAR406 clade), all usually being the most ubiquitous free-living bacterial lineages and dominantly distributed in epi- and mesopelagic zones (Grote et al., 2012; Tarn et al., 2016; Yilmaz et al., 2016; Milici et al., 2017; Liu et al., 2018a). Genomic information suggests that, although these clades have a flexible metabolism utilizing multiple hydrocarbon compounds, they generally lack carbohydrate-active enzyme genes for the attachment to and the degradation of particulate organic matter (Peoples et al., 2018), consistent with their preference for the free-living lifestyle rather than particle attachment (Eloe et al., 2011; Salazar et al., 2015; Tarn et al., 2016). In addition, the percentages of the SAR11 clade revealed here seem to be relatively lower compared with those reported in previous studies where the SAR11 clade typically makes up 20 % to 40 % of the bacterioplankton (Morris et al., 2002; Apprill et al., 2015). It may be related to the sequencing primers used, which potentially cause underestimation of the SAR11 clade and bias the interpretation of their relative abundances (Apprill et al., 2015).
In addition to those predominant lineages mentioned above, there are a couple of bacterial taxa showing evident PA or FL preferences. At the family level, these PA- or FL-preferred taxa are indicated well by their odds ratio between PA and FL fractions. These bacterial lineages are characterized by low abundances or occasional occurrences in water columns (Fig. 6, Table S1) but a high odds ratio (absolute value) (Fig. 8), indicating their strong preferential divergence in the two size fractions. The majority of these lineages are recorded consistently regarding their PA or FL preferences in previous studies and commonly possess the ability to hydrolyze and utilize complex carbon sources. Although their abundance is low, these relatively minor populations can still effectively influence local microhabitats because of their high specificity for organics. In contrast, there are still some populations which are scarcely reported. For example, the Sva0996 marine group, an actinobacterial group, is retrieved occasionally from marine sediments and the upper ocean (Bano and Hollibaugh, 2002; Wang et al., 2018). Orsi et al. (2016) first found this group prefers the free-living lifestyle in upper seawater and has the ability to assimilate phytoplankton-derived dissolved protein. Our present results suggest that the Sva0996 group is flexible in adapting to PA or FL lifestyles at the surface seawater because the two lifestyles occur concurrently. Moreover, the distribution of the Sva0996 group is not restricted only in the upper photic ocean, and they can survive in meso- and bathypelagic seawaters with the significant preference for the free-living lifestyle (Fig. 8). However, due to a lack of pure culture or their genome information, it is not yet possible to elaborate on their preferences for PA and FL lifestyles.
A high proportion of bacterial lineages are revealed to co-occur in both PA and FL fractions (Figs. 8 and S7), indicating that a considerable amount of bacterial lineages potentially have PA and FL dual-lifestyle strategies. On the one hand, as shown in Fig. 6, a few bacterial lineages co-occur in PA and FL fractions at least at one of the same depths with approximately equivalent abundances. In such cases, their odds ratios are close to zero or minor range (Fig. 8), indicating that these bacteria are able to employ two different survival strategies at the same time. On the other hand, lots of taxa show divergent preferences for PA or FL lifestyles at different depths or different locations. This is clearly evident by the shift or conversion of their odds ratios at different depths along the vertical profiles of the water column (Fig. 8), which is indicative of their different adaption tactics to different environments. One possible explanation is that most of the marine bacteria are generalists with dual-lifestyle strategies (Bauer et al., 2006; Gonzalez et al., 2008) and able to grow in suspension as well as on particles (Lee et al., 2004; Grossart et al., 2006, 2010). For instance, PA bacteria must be capable of surviving freely in the water column to migrate and colonize new organic particles (Ghiglione et al., 2007; Crespo et al., 2013). Bacterial populations may switch their lifestyles between free-living and particle-attachment, depending on substrate availability and the surrounding chemical triggers (Grossart, 2010; D'Ambrosio et al., 2014). To date, one exception, the genus Scalindua in the Planctomycetes phylum, which is a known marine chemoautotroph involved in anammox, is exclusively observed in FL fractions in previous studies (Fuchsman et al., 2012; Ganesh et al., 2014; Suter et al., 2018). However, it is absent from our water columns.
4.4 Archaeal community preferences for PA and FL lifestyles
Samples of PA and FL archaeal fractions were also separated into different groups by statistical analysis (Fig. 3b, Table S3), indicating their different community structures. However, because most of OTUs belonged to uncultured archaeon, it is impossible to assign them into taxonomic lineages at a finer level. Thus, the distinction of archaeal population compositions between PA and FL fractions was unnoticeable (Fig. 7). The Nitrosopumilales under MGI and MGII are the most abundant taxa in both PA and FL archaeal fractions. The thaumarchaeotal Nitrosopumilales are one of the most abundant and cosmopolitan chemolithoautotrophs in the dark ocean (Konneke et al., 2005) and responsible for much of the ammonia oxidation in this environment for their common metabolism of aerobic ammonia oxidation. Corresponding to their autotrophic metabolism, MGI (including Nitrosopumilales) generally exhibit a free-living preference and are the prevalent archaeal taxa in free-living fractions below euphotic zone (Smith et al., 2013; Salazar et al., 2015; Tarn et al., 2016). However, different from our results, a few studies showed that MGI dominated both the PA and FL archaeal populations, and no obvious distinction was observed in abundance and ecotype of MGI (Eloe et al., 2011; Jing et al., 2018). To date, only a few pure cultures of marine MGI, small rods with a diameter of 0.15–0.26 µm, and a length of 0.5–1.59 µm and no flagella were observed (Könneke et al., 2005; Qin et al., 2014), suggesting that their occurrence in the PA fraction is not caused by the pore size of the filter to fractionate different assemblages. One possibility is that decomposition of organic particles continuously releases ammonia, and MGI can easily acquire high concentrations of ammonia by attaching to particles, especially in an oligotrophic area. Recent studies provide another explanation for the particle-attached MGI: that some MGI cultures are obligate mixotrophs that rely on uptake and assimilation of organic compounds (Alonso-Sáez et al., 2012; Qin et al., 2014). In such a case, the PA lifestyle is in favor of their nutrient requirements. MGII have a wide distribution in the open ocean, and as shown in our results, they are the dominant archaeal community generally within the upper euphotic zone (Massana et al., 2000; Martin-Cuadrado et al., 2015). Recently, they have been found, however, to be also abundant in deep-sea waters (Baker et al., 2013; Tarn et al., 2016; Liu et al., 2018a), showing a wider adaption to diverse marine habitats in addition to the photic zone. MGII are thought to be heterotrophs and have the ability of degrading proteins and lipids (Iverson et al., 2012; Orsi et al., 2015). Metagenomes revealed a number of genes encoding cell adhesion, degradation of high-molecular-weight organic matter and photoheterotrophy (Rinke et al., 2019; Tully et al., 2019), evidencing their potential to utilize organic particles as important growth substrates. All these findings imply MGII's preference for the particle-attached lifestyle, and they are frequently detected from PA fractions in size-fractionated studies (Iverson et al., 2012; Orsi et al., 2015; Tarn et al., 2016). However, in a few studies including our present study, MGII are also identified as the dominant archaeal components from FL fractions, with equal or even more abundance than PA fractions (Fig. 7). Further studies confirm that genome contents and populations of free-living MGII are distinct from those of particle-attached MGII (Orsi et al., 2015; Rinke et al., 2019), suggesting their metabolic evolution and adjustment to niche partitioning. In addition, MGIII also occurred commonly in both fractions (Fig. 7). MGIII are usually retrieved as minor components of deep mesopelagic and bathypelagic communities (Galand et al., 2009; Tarn et al., 2016). Like MGII, to date no cultured representative of MGIII has led to the result that little is known about their ecological and physiological characteristics. Function prediction from metagenomes suggests that MGIII are aerobic (or facultative anaerobic), motile, and heterotrophic and potentially can utilize lipid, proteins and polysaccharides as major energy sources (Martin-Cuadrado et al., 2008; Haro-Moreno et al., 2017). Recently, a novel lineage of MGIII genomes preferring to live in the photic zone was recovered, consistent with few previous studies and our present results in which MGIII populations are obtained from the euphotic zone with a considerable abundance (Galand et al., 2009, 2010). Moreover, recent findings also indicate that MGIII are inclined to be attached to other microorganisms (particle-attached preference) and only sporadically be released to the surrounding environments (free-living lifestyle) (Haro-Moreno et al., 2017).
In addition, there are several other archaeal lineages with remarkable differences in abundance between PA and FL fractions. The orders Methanosarcinales and Methanobacteriales, affiliated with the phylum Euryarchaeota and retrieved exclusively from PA fractions (Fig. 7), belong to strictly anaerobic methanogens. Their preference for the particle-attached lifestyle in the water column is intelligible. Within the normal water column, seawater is usually oxic in spite of low oxygen concentration. Only on or inside the organic particles where heterotrophic microbes attach and digest organic matter using oxygen as an electron acceptor, local anoxic niches are developed with the gradual exhaustion of ambient oxygen and become suitable for the survival of anaerobic methanogens. Members of the Woesearchaeota were abundantly derived from the PA fraction of the upper seawater. In marine environments, Woesearchaeota are distributed restrictively in marine sediments (Lipsewers et al., 2018) or deep-sea hydrothermal vents (Takai and Horikoshi, 1999) and are scarcely detected from pelagic seawater masses. Recent studies suggest that woesearchaeotal lineages are mostly retrieved from anoxic environments (Castelle et al., 2015; Liu et al., 2018b). Moreover, genomic metabolic analysis indicates Woesearchaeota have an anaerobic heterotrophic lifestyle with conspicuous metabolic deficiencies (Probst et al., 2017; Liu et al., 2018b), implying a potential syntrophic or mutualistic partnership with other organisms (Castelle et al., 2015; Liu et al., 2018b). It is further demonstrated that Woesearchaeota tend to co-occur with typical anaerobic methanogens from the Methanomicrobia and Methanobacteria, constituting a potential consortia (Liu et al., 2018b). In our present results, at several depths, the Methanosarcinales of the Methanomicrobia and the Methanobacteriales of the Methanobacteria, together with Woesearchaeota, were detected concurrently, implying to a large extent their potential syntrophic partnership.
4.5 Potential vertical connectivity of microbial populations along the depth profile
Microbial distribution at different depths to a certain extent implicates their potential vertical connectivity along the water column profile. It has been suggested that the sinking of organic particles formed in the upper euphotic zone is a main vector in transferring prokaryotes from the surficial ocean to deep waters (Mestre et al., 2018). Those surficial lineages, usually belonging to putative photosynthetic/photoheterotrophic, bacteriochlorophyll a-containing microorganism or strict epipelagic/euphotic inhabitants, are reliable indicators of their downward transportation if they are detected from meso- or bathypelagic waters. For example, cyanobacteria are typical photosynthetic bacteria, and their distribution is thought to be confined to the euphotic zone, with commonly observed maximum depths of about 150–200 m. In the present study, however, cyanobacterial lineages were retrieved throughout the whole water column (Figs. 5 and 6), especially at 4000 m depth where cyanobacteria account for nearly 12 % of the PA communities. Although a recent study revealed that cyanobacteria can dominate the deep continental subsurface microbial communities with the potential for a hydrogen-based lithoautotrophic metabolism instead of photosynthesis (Puente-Sanchez et al., 2018), these indigenous deep cyanobacteria were classified into the genera Calothrix, Microcoleus and Chroococcidiopsis, which is phylogenetically different from those prevailing in our study (Prochlorococcus, Synechococcus). Jiao et al. (2014) observed substantial Prochlorococcus populations at 1500 m depth in the South China Sea and suggested that multiple physical processes, including internal solitary waves and mesoscale eddies, were responsible for the occurrence of these “deep Prochlorococcus”. However, in our study area, ages of seawater increase gradually from the surface to the deep along the water column profile in a normal time sequence (Table 1), refuting this possibility. Thus, a reasonable postulation here is that the sinking particles function as vectors and convey cyanobacteria attaching to particle surfaces from the epipelagic zone into deep-sea waters. Likewise, members of the family Erythrobacteraceae, which are largely represented by OTUs within the genus Erythrobacter, are also present abundantly in both PA and FL fractions at 4000 m depth (Fig. 6). Erythrobacter spp. belong to putative bacteriochlorophyll a-containing, aerobic anoxygenic photoheterotrophic bacteria and are thought to be distributed only in the euphotic upper ocean (Kolber et al., 2000; Koblížek et al., 2003). Members of the SAR11 clade are potentially photoheterotrophic (Gomez-Pereira et al., 2013; Evans et al., 2015) and ubiquitous in global photic zones as one of the most abundant bacteria (Morris et al., 2002). We observed that members of the SAR11 clade are distributed across the whole water columns, especially in mesopelagic aphotic depths with relatively high proportions. Other lineages specializing in inhabiting surface seawater but also retrieved from the deep ocean include the gammaproteobacterial SAR86 clade, the SAR116 clade of marine Roseobacter and the SAR202 clade within Chloroflexi. The majority of the OTUs within these surface lineages have been retrieved from the meso-/bathypelagic ocean and can be traced back simultaneously to those present in surface waters, suggesting their potential origin from the upper epipelagic zones.
In this study, we systematically compared bacterial and archaeal community structures within two different filtration fractions representing particle-attached and free-living lifestyles at different depths in the South China Sea. As revealed in previous studies, for either bacteria or archaea, the FL fractions usually show higher cell abundance and diversity than their PA counterparts at most depths. A set of environmental factors including depth, salinity, seawater age, DOC, POC, DO and silicate are considered to play important roles in structuring PA and FL microbial communities along the depth profile. On the one hand, as the result of adapting to different organic substrates available, PA and FL fractions generally accommodate significantly divergent microbial compositions at each depth. At fine taxonomic levels, a considerable number of microbial lineages exhibited pronounced preferences for PA or FL lifestyles, also with distinct stratified distribution along the depth profile. A few microbial taxa show potentially PA and FL dual-lifestyle strategies, able to switch according to substrate availability and environmental variations, implying versatile metabolic flexibility. In addition, we found that the sinking organic particles likely function as vectors in prokaryote transfer from the surface ocean to deep waters, indicating the potential vertical connectivity of prokaryotes along the water column profile.
The pyrosequencing data obtained from the 454 sequencing of 16S rRNA genes were deposited in the Sequence Read Archive (SRA) database under accession ID PRJNA546072 for bacterial sequences and PRJNA546071 for archaeal sequences.
The supplement related to this article is available online at: https://doi.org/10.5194/bg-18-113-2021-supplement.
JL and JF designed the experiments, and JL, LG, JW and BW carried them out. JL, SB, LZ and LS treated and analyzed the sequence data. JL and JF wrote the manuscript with contributions from all co-authors.
The authors declare that they have no conflict of interest.
We thank all the crew and scientists on the open cruise of R/V Dongfanghong II in July 2014. We would also like to thank three anonymous reviewers for their valuable comments.
This research has been supported by the National Natural Science Foundation of China (grant no. 41373071 and 91951210) and the National Key R&D Program of China (grant no. 2018YFC0310600).
This paper was edited by Carol Robinson and reviewed by three anonymous referees.
Acinas, S. G., Rodríguez-Valera, F., and Pedrós-Alió, C.: Spatial and temporal variation in marine bacterioplankton diversity as shown by RFLP fingerprinting of PCR amplified 16S rDNA, FEMS Microbiol. Ecol., 24, 27–40, 1997.
Alldredge, A. L., Cole, J. J., and Caron, D. A.: Production of heterotrophic bacteria inhabiting macroscopic organic aggregates (marine snow) from surface waters, Limnol. Oceanogr., 31, 68–78, 1986.
Alonso-Sáez, L., Waller, A. S., Mende, D. R., Bakker, K., Farnelid, H., Yager, P. L., Lovejoy, C., Tremblay, J.-É., Potvin, M., Heinrich, F., Estrada, M., Riemann, L., Bork, P., Pedrós-Alió, C., and Bertilsson, S.: Role for urea in nitrification by polar marine Archaea, P. Natl. Acad. Sci. USA, 109, 17989–17994, https://doi.org/10.1073/pnas.1201914109, 2012.
Apprill, A., McNally, S., Parsons, R., and Weber, L.: Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton, Aquat. Microb. Ecol., 75, 129–137, https://doi.org/10.3354/ame01753, 2015.
Arístegui, J., Gasol, J. M., Duarte, C. M., and Herndl, G. J.: Microbial oceanography of the dark ocean's pelagic realm, Limnol.Oceanogr., 54, 1501–529, 2009.
Azam, F. and Malfatti, F.: Microbial structuring of marine ecosystems, Nat. Rev. Microbiol., 5, 782–791, 2007.
Baker, B. J., Sheik, C. S., Taylor, C. A., Jain, S., Bhasi, A., Cavalcoli, J. D., and Dick, G. J.: Community transcriptomic assembly reveals microbes that contribute to deep-sea carbon and nitrogen cycling, ISME J., 7, 1962–973, https://doi.org/10.1038/ismej.2013.85, 2013.
Bano, N. and Hollibaugh, J. T.: Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean, Appl. Environ. Microbiol., 68, 505–518, 2002.
Bano, N., Ruffin, S., Ransom, B., and Hollibaugh, J. T.: Phylogenetic composition of Arctic Ocean archaeal assemblages and comparison with antarctic assemblages, Appl. Environ. Microbiol., 70, 781–789, 2004.
Bauer, M., Kube, M., Teeling, H., Richter, M., Lombardot, T., Allers, E., Wurdemann, C. A., Quast, C., Kuhl, H., Knaust, F., Woebken, D., Bischof, K., Mussmann, M., Choudhuri, J. V., Meyer, F., Reinhardt, R., Amann, R. I., and Glockner, F. O.: Whole genome analysis of the marine Bacteroidetes “Gramella forsetii” reveals adaptations to degradation of polymeric organic matter, Environ. Microbiol., 8, 2201–2213, 2006.
Cammen, L. M. and Walker, J. A.: Distribution and activity of attached and free-living suspended bacteria in the bay of fundy, Can. J. Fish. Aquat. Sci., 39, 1655–1663, 1982.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., Fierer, N., Peña, A. G., Goodrich, J. K., Gordon, J. I., Huttley, G. A., Kelley, S. T., Knights, D., Koenig, J. E., Ley, R. E., Lozupone, C. A., McDonald, D., Muegge, B. D., Pirrung, M., Reeder, J., Sevinsky, J. R., Turnbaugh, P. J., Walters, W. A., Widmann, J., Yatsunenko, T., Zaneveld, J., and Knight, R.: QIIME allows analysis of high-throughput community sequencing data, Nat. Methods., 7, 335–336, https://doi.org/10.1038/nmeth.f.303, 2010.
Caron, D. A., Davis, P. G., Madon, L. P., and Sieburth, J. M.: Heterotrophic bacteria and bacteriovorous protozoa in oceanic macroaggregates, Science, 218, 795–797, 1982.
Castelle, C., Wrighton, K., Thomas, B., Hug, L., Brown, C., Wilkins, M., Frischkorn, K. R., Tringe, S. G., Singh, A., Markillie, L. M., Taylor, R. C., Williams, K. H., and Banfield, J. F.: Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling, Curr. Biol., 25, 690–701, 2015.
Chen, M., Liu, H., Song, S., and Sun, J.: Size-fractionated mesozooplankton biomass and grazing impact on phytoplankton in northern South China Sea during four seasons, Deep-Sea Res. Pt. II., 117, 108–118, 2015.
Chen, W., Cai, P., Dai, M., and Wei, J.: 234Th ∕ 238U disequilibrium and particulate organic carbon export in the northern South China Sea, J. Oceanogr., 64, 417–428, 2008.
Chen, Y. L., Chen, H. Y., Karl, D. M., and Takahashi, M.: Nitrogen modulates phytoplankton growth in spring in the South China Sea, Cont. Shelf Res., 24, 527–541, 2004.
Cram, J. A., Weber, T., Leung, S. W., McDonnell, A. M. P., Liang, J. H., and Deutsch, C.: The role of particle size, ballast, temperature, and oxygen in the sinking flux to the deep sea, Global Biogeochem. Cy., 32, 858–876, 2018.
Crespo, B. G., Pommier, T., Fernadez-Gomez, B., and Pedros-Alio, C.: Taxonomic composition of the particle-attached and free-living bacterial assemblages in the Northwest Mediterranean Sea analyzed by pyrosequencing of the 16S rRNA, Microbiology Open, 2, 541–552, 2013.
Crump, B. C., Baross, J. A., and Simenstad, C. A.: Dominance of particle-attached bacteria in the Columbia River estuary, USA, Aquat. Microb. Ecol., 14, 7–18, 1998.
D'Ambrosio, L., Ziervogel, K., MacGregor, B., Teske, A., and Arnosti, C.: Composition and enzymatic function of particle-associated and free-living bacteria: a coastal/offshore comparison, ISME J., 8, 2167–2179, https://doi.org/10.1038/ismej.2014.67, 2014.
DeLong, E. F., Franks, D. G., and Alldredge, A. L.: Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages, Limnol. Oceanogr., 38, 924–934, 1993.
Dilly, O., Bloem, J., Vos, A., and Munch, J. C.: Bacterial diversity in agricultural soils during litter decomposition, Appl. Environ. Microbiol., 70, 468–474, 2004.
Doxaran, D., Ehn, J., Bélanger, S., Matsuoka, A., Hooker, S., and Babin, M.: Optical characterisation of suspended particles in the Mackenzie River plume (Canadian Arctic Ocean) and implications for ocean colour remote sensing, Biogeosciences, 9, 3213–3229, https://doi.org/10.5194/bg-9-3213-2012, 2012.
Eloe, E. A., Shulse, C. N., Fadrosh, D. W., Williamson, S. J., Allen, E. E., and Bartlett, D. H.: Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment, Env. Microbiol. Rep., 3, 449–458, 2011.
Evans, P. N., Parks, D. H., Chadwick, G. L., Robbins, S. J., Orphan, V. J., and Golding, S. D., and Tyson, G. W.: Methane metabolism in the archaeal phylum bathyarchaeota revealed by genome-centric metagenomics, Science, 350, 434–438, 2015.
Fuchsman, C. A., Staley, J. T., Oakley, B. B., Kirkpatrick, J. B., and Murray, J. W.: Free-living and aggregate-associated Planctomycetes in the Black Sea, FEMS Microbiol. Ecol., 80, 402–416, 2012.
Fuhrman, J. A. and Davis, A. A.: Widespread Archaea and novel Bacteria from the deep sea as shown by 16S rRNA gene sequences, Mar. Ecol. Prog. Ser., 150, 275–285, 1997.
Galand, P. E., Lovejoy, C., Pouliot, J., and Vincent, W. F.: Heterogeneous archaeal communities in the particle rich environment of an arctic shelf ecosystem, J. Mar. Syst., 74, 774–782, 2008.
Galand, P. E., Casamayor, E. O., Kirchman, D. L., Potvin, M., and Lovejoy, C.: Unique archaeal assemblages in the Arctic Ocean unveiled by massively parallel tag sequencing, ISME J., 3, 860–869, 2009.
Galand, P. E., Gutiérrez-Provecho, C., Massana, R., Gasol, J. M., and Casamayor, E. O.: Inter-annual recurrence of archaeal assemblages in the coastal NW Mediterranean Sea (Blanes Bay microbial observatory), Limnol. Oceanogr., 55, 2117–2125, 2010.
Garneau, M.È., Vincent, W. F., Terrado, R., and Lovejoy, C.: Importance of particle-associated bacterial heterotrophy in a coastal Arctic ecosystem, J. Mar. Syst., 75, 185–197, 2009.
Ganesh, S., Parris, D. J., DeLong, E. F., and Stewart, F. J.: Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone, ISME J., 8, 187–211, https://doi.org/10.1038/ismej.2013.144, 2014.
Ghai, R., Mizuno, C. M., Picazo, A., Camacho, A., and Rodriguez-Valera, F.: Metagenomics uncovers a new group of low GC and ultra-small marine Actinobacteria, Sci. Rep., 3, 2471, https://doi.org/10.1038/srep02471, 2013.
Ghiglione, J. F., Mevel, G., Pujo-Pay, M., Mousseau, L., Lebaron, P., and Goutx, M.: Diel and seasonal variations in abundance, activity, and community structure of particle-attached and free-living bacteria in NW Mediterranean Sea, Microb. Ecol., 54, 217–231, 2007.
Ghiglione, J. F., Conan, P., and Pujo-Pay, M.: Diversity of total and active free-living vs. particle-attached bacteria in the euphotic zone of the NW Mediterranean Sea, FEMS Microbiol. Lett., 299, 9–21, 2009.
Giovannoni, S. J. and Stingl, U.: Molecular diversity and ecology of microbial plankton, Nature, 437, 343–348, https://doi.org/10.1038/nature04158, 2005.
Giovannoni, S. J., Cameron, Thrash J., and Temperton, B.: Implications of streamlining theory for microbial ecology, ISME J., 8, 1553–1565, https://doi.org/10.1038/ismej.2014.60, 2014.
Gómez-Pereira, P. R., Kennaway, G., Fuchs, B. M., Tarran, G. A., and Zubkov, M. V.: Flow cytometric identification of Mamiellales clade II in the Southern Atlantic Ocean, FEMS Microbiol. Ecol., 83, 664–671, 2013.
Gong, G. C., Liu, K. K., Liu, C. T., Pai, and S. C.: The chemical hydrography of the South China Sea west of Luzon and a comparison with the West Philippine Sea, Terr. Atmos. Ocean Sci., 3, 587–602, 1992.
Gonzalez, J. M., Fernandez-Gomez, B., Fernandez-Guerra, A., Gomez-Consarnau, L., Sanchez, O., and Coll-Llado, M.: Genome analysis of the proteorhodopsin-containing marine bacterium Polaribacter sp. MED152 (Flavobacteria), P. Natl. Acad. Sci. USA, 105, 8724–8729, 2008.
Green, P. N.: Methylobacterium, in: The Prokaryotes: A Handbook on the Biology of Bacteria, edited by: Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K. H., and Stackebrandt, E., Springer, New York, 5, 257–265, 2006.
Griffith, P., Shiah, F., Gloersen, K., Ducklow, H. W., and Fletcher, M.: Activity and distribution of attached bacteria in Chesapeake Bay, Mar. Ecol. Prog. Ser., 108, 1–10, 1994.
Grossart, H. P.: Ecological consequences of bacterioplankton lifestyles: changes in concepts are needed, Env. Microbiol. Rep., 2, 706–714, 2010.
Grossart, H. P., Kiorboe, T., Tang, K., Allgaier, M., Yam, E. M., and Ploug, H.: Interactions between marine snow and heterotrophic bacteria: aggregate formation and microbial dynamics, Aquat. Microb. Ecol., 42, 19–26, 2006.
Grossart, H.-P., Tang, K. W., Kiørboe, T., and Ploug, H.: Comparison of cell-specific activity between free-living and attached bacteria using isolates and natural assemblages, FEMS Microbiol. Lett., 266, 194–200, 2007.
Grote, J., Thrash, J. C., Huggett, M. J., Landry, Z. C., Carini, P., Giovannoni, S. J., and Rappé, M. S.: Streamlining and core genome conservation among highly divergent members of the SAR11 clade, mBio, 3, e00252-12, https://doi.org/10.1128/mBio.00252-12, 2012.
Haro-Moreno, J. M., Rodriguez-Valera, F., López-García, Purificación, Moreira, D., and Martin-Cuadrado, A. B.: New insights into marine group III Euryarchaeota, from dark to light, ISME J., 11, 1102–1117, 2017.
He, X., Xu, D., Bai, Y., Pan, D., Chen, T. A., Chen, X., and Gong, F.: Eddy-entrained Pearl River plume into the oligotrophic basin of the South China Sea, Cont. Shelf Res., 124, 117–124, 2016.
Hollibaugh, J. T., Wong, P. S., and Murrell, M. C.: Similarity of particle-associated and free-living bacterial communities in northern San Francisco Bay, California, Aquat. Microb. Ecol., 21, 103–114, 2000.
Huang, Y. N., Chen, F. J., Zhao, H., Zeng, Z., and Chen, J. F.: Concentration distribution and structural features of nutrients in the northwest of South China Sea in winter 2012, J. Appl. Oceanogr., 34, 310–316, 2015.
Iverson, V., Morris, R. M., Frazar, C. D., Berthiaume, C. T., Morales, R. L., and Armbrust, E. V.: Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota, Science, 335, 587–590, 2012.
Jiao, N., Luo, T., Zhang, R., Yan, W., Lin, Y., Johnson, Z. I., Tian, J., Yuan, D., Yang, Q., Zheng, Q., Sun, J., Hu, D., and Wang, P.: Presence of Prochlorococcus in the aphotic waters of the western Pacific Ocean, Biogeosciences, 11, 2391–2400, https://doi.org/10.5194/bg-11-2391-2014, 2014.
Jing, H., Zhu, W., Liu, H., Zheng, L., and Zhang, Y.: Particle-attached and free-living archaeal communities in the benthic boundary layer of the Mariana Trench, Front. Microbiol., 9, 2821, https://doi.org/10.3389/fmicb.2018.02821, 2018.
Karner, M. and Herndl, G. J.: Extracellular enzymatic activity and secondary production in free-living and marine-snow-associated bacteria, Mar. Biol., 113, 341–347, 1992.
Karner, M. B., DeLong, E. F., and Karl, D. M.: Archaeal dominance in the mesopelagic zone of the Pacific Ocean, Nature, 409, 507–510, 2001.
Kiorboe, T. and Jackson, G. A.: Marine snow, organic solute plumes, and optimal chemosensory behavior of bacteria, Limnol. Oceanogr., 46, 1309–1318, 2001.
Koblížek, M., Béjà, O., Bidigare, R. R., Christensen, S., Benitez-Nelson, B., Vetriani, C., Kolber, M. K., Falkowski, P. G., and Kolber, Z. S.: Isolation and characterization of Erythrobacter sp. strains from the upper ocean, Arch. Microbiol., 180, 327–338, https://doi.org/10.1007/s00203-003-0596-6, 2003.
Kolber, Z. S., Van Dover, C. L., Niederman, R. A., and Falkowski, P. G.: Bacterial photosynthesis in surface waters of the open ocean, Nature, 407, 177–179, 2000.
Könneke M, Bernhard, A. E., de la Torre, J. R., Walker, C. B., Waterbury, J. B., and Stahl, D. A.: Isolation of an autotrophic ammonia-oxidizing marine archaeon, Nature, 437, 543–546, 2005.
Laufkötter, C., John, J. G., Stock, C. A., and Dunne, J. P.: Temperature and oxygen dependence of the remineralization of organic matter, Global Biogeochem. Cy., 31, 1038–1050, 2017.
Lauro, F. M., McDougald, D., Thomas, T., Williams, T. J., Egan, S., Rice, S., DeMaere, M. Z., Ting, L., Ertan, H., Johnson, J., Ferriera, S., Lapidus, A., Anderson, I., Kyrpides, N., Munk, A. C., Detter, C., Han, C. S., Brown, M. V., Robb, F. T., Kjelleberg, S., and Cavicchioli, R.: The genomic basis of trophic strategy in marine bacteria, P. Natl. Acad. Sci. USA, 106, 15527–15533, https://doi.org/10.1073/pnas.0903507106, 2009.
Lee, C., Wakeham, S., and Arnosti, C.: Particulate organic matter in the sea: the composition conundrum, Ambio, 33, 566–575, 2004.
Lee, Z. M.-P., Bussema, C., and Schmidt, T. M.: rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea, Nucl. Acid. Res., 37, 489–493, 2009.
Li, J., Wei, B., Wang, J., Liu, Y., Dasgupta, S., and Zhang, L.: Variation in abundance and community structure of particle-attached and free-living bacteria in the South China Sea, Deep-Sea Res. Pt. II, 122, 64–73, https://doi.org/10.1016/j.dsr2.2015.07.006, 2015.
Liang, W., Tang, D., and Luo, X.: Phytoplankton size structure in the western South China Sea under the influence of a “jet-eddy system”, J. Mar. Syst., 187, 82–95, 2018.
Lipsewers, Y. A., Hopmans, E. C., Sinninghe Damsté, J. S., and Villanueva, L.: Potential recycling of thaumarchaeotal lipids by DPANN Archaea in seasonally hypoxic surface marine sediments, Org. Geochem., 119, 101–109, 2018.
Liu, R. L., Wang, L., Liu, Q. F., Wang, Z. X., Li, Z. Z., and Fang, J. S.: Depth-resolved distribution of particle-attached and free-living bacterial communities in the water column of the New Britain Trench, Front. Microbiol., 9, 625, https://doi.org/10.3389/fmicb.2018.00625, 2018a.
Liu, S., Riesen, A., and Liu, Z.: Differentiating the role of different-sized microorganisms in peptide decomposition during incubations using size-fractioned coastal seawater, J. Exp. Mar. Biol. Ecol., 472, 97–106, 2015.
Liu, X., Li, M., Castelle, C. J., Probst, A. J., Zhou, Z., and Pan, J.: Insights into the ecology, evolution, and metabolism of the widespread woesearchaeotal lineages, Microbiome, 6, 102, https://doi.org/10.1186/s40168-018-0488-2, 2018b.
Long, R. A. and Azam, F.: Microscale patchiness of bacterioplankton assemblage richness in seawater, Aquat. Microb. Ecol., 26, 103–113, https://doi.org/10.3354/ame026103, 2001.
Martin-Cuadrado, A.-B., Rodriguez-Valera, F., Moreira, D., Alba, J. C., Ivars-Martinez, E., and Henn, M. R.: Hindsight in the relative abundance, metabolic potential and genome dynamics of uncultivated marine archaea from comparative metagenomic analyses of bathypelagic plankton of different oceanic regions, ISME J., 2, 865–886, 2008.
Martin-Cuadrado, A.-B., Garcia-Heredia, I., Moltó, A.G., López-Úbeda, R., Kimes, N., López-García, P., Moreira, D., and Rodriguez-Valera, F.: A new class of marine euryarchaeota group II from the Mediterranean deep chlorophyll maximum, ISME J., 9, 1619–1634, https://doi.org/10.1038/ismej.2014.249, 2015.
Massana, R., Delong, E. F., and Pedros-Alio, C.: A few cosmopolitan phylotypes dominate planktonic archaeal assemblages in widely different oceanic provinces, Appl. Environ. Microbiol., 66, 1777–1787, 2000.
Meng, F., Dai, M., Cao, Z., Wu, K., Zhao, X., Li, X., Chen, J., and Gan, J.: Seasonal dynamics of dissolved organic carbon under complex circulation schemes on a large continental shelf: the northern South China Sea, J. Geophys. Res.-Ocean., 122, 9415–9428, 2017.
Mestre, M., Ruiz-Gonzalez, C., Logares, R., Duarte, C. M., Gasol, J. M., and Sala, M. M.: Sinking particles promote vertical connectivity in the ocean microbiome, P. Natl. Acad. Sci. USA, 115, 6799–6807, 2018.
Mevel, G., Vernet, M., Goutx, M., and Ghiglione, J. F.: Seasonal to hour variation scales in abundance and production of total and particle-attached bacteria in the open NW Mediterranean Sea (0–1000 m), Biogeosciences, 5, 1573–1586, https://doi.org/10.5194/bg-5-1573-2008, 2008.
Milici, M., Deng, Z. L., Tomasch, J., Decelle, J., Wos-Oxley, M. L., Wang H., Jáuregui, R., Plumeier, I., Giebel, H. A., Badewien, T. H., Wurst, M., Pieper, D. H., Simon, M., and Wagner-Döbler, I.: Co-occurrence analysis of microbial taxa in the Atlantic Ocean reveals high connectivity in the free-living bacterioplankton, Front. Microbiol., 7, 649, https://doi.org/10.3389/fmicb.2016.00649, 2016.
Milici, M., Vital, M., Tomasch, J., Badewien, T. H., Giebel, H. A., and Plumeier, I.: Diversity and community composition of particle-associated and free-living bacteria in mesopelagic and bathypelagic Southern Ocean water masses: evidence of dispersal limitation in the Bransfield Strait, Limnol. Oceanogr., 62, 1080–1095, https://doi.org/10.1002/lno.10487, 2017.
Mincer, T. J., Church, M. J., Taylor, L. T., Preston, C., Karl, D. M., and DeLong, E. F.: Quantitative distribution of presumptive archaeal and bacterial nitrifiers in Monterey Bay and the North Pacific Subtropical Gyre, Environ. Microbiol., 9, 1162–1175, 2007.
Mizuno, C. M., Rodriguez-Valera, F., and Ghai, R.: Genomes of planktonic acidimicrobiales: widening horizons for marine actinobacteria by metagenomics, mBio, 6, e02083-14, https://doi.org/10.1128/mBio.02083-14, 2015
Moeseneder, M. M., Winter, C., and Herndl, G. J.: Horizontal and vertical complexity of attached and free-living bacteria of the eastern Mediterranean Sea, determined by 16S rDNA and 16S rRNA fingerprints, Limnol. Oceanagr., 46, 95–107, 2001.
Mönnich, J., Tebben, J., Bergemann, J., Case, R., Wohlrab, S., and Harder, T.: Niche-based assembly of bacterial consortia on the diatom Thalassiosira rotula is stable and reproducible, ISME J., 14, 1614–1625, https://doi.org/10.1038/s41396-020-0631-5, 2020.
Morris, S. A., Radajewski, S., Willison, T. W., and Murrell, J. C.: Identification of the functionally active methanotroph population in a peat soil microcosm by stable-isotope probing, Appl. Environ. Microbiol., 68, 1446–1453, 2002.
Ohene-Adjei, S., Teather, R. M., Ivan, M., and Forster, R. J.: Postinoculation protozoan establishment and association patterns of methanogenic Archaea in the ovine rumen, Appl. Environ. Microbiol., 73, 4609–4618, 2007.
Orsi, W. D., Smith, J. M., Wilcox, H. M., Swalwell, J. E., Carini, P., and Worden, A. Z.: Ecophysiology of uncultivated marine euryarchaea is linked to particulate organic matter, ISME J., 9, 1747–1763, 2015.
Orsi, W. D., Smith, J. M., Liu, S., Liu, Z., Sakamoto, C. M., Wilken, S., Poirier, C., Richards, T. A., Keeling, P. J., Worden, A. Z., and Santoro, A. E.: Diverse, uncultivated bacteria and archaea underlying the cycling of dissolved protein in the ocean, ISME J., 10, 2158–2173, https://doi.org/10.1038/ismej.2016.20, 2016.
Ortega-Retuerta, E., Joux, F., Jeffrey, W. H., and Ghiglione, J. F.: Spatial variability of particle-attached and free-living bacterial diversity in surface waters from the Mackenzie River to the Beaufort Sea (Canadian Arctic), Biogeosciences, 10, 2747–2759, https://doi.org/10.5194/bg-10-2747-2013, 2013.
Peoples, L. M., Sierra, D., Oladayo, O., Qing, X., Alex, N., and Jessica, B.: Vertically distinct microbial communities in the Mariana and Kermadec Trenches, PLOS ONE, 13, e0195102, https://doi.org/10.1371/journal.pone.0195102, 2018.
Poretsky, R. S., Sun, S., Mou, X., and Moran, M. A.: Transporter genes expressed by coastal bacterioplankton in response to dissolved organic carbon, Environ. Microbiol., 12, 616–627, 2010.
Probst, A. J., Castelle, C. J., Singh, A., Brown, C. T., Anantharaman, K., Sharon, I., Hug, L. A., Burstein, D., Emerson, J. B., Thomas, B. C., and Banfield, B. F.: Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations, Environ. Microbiol., 19, 459–74, 2017.
Puente-Sánchez, F., Arce-Rodríguez, A., Oggerin, M., García-Villadangos, M., Moreno-Paz, M., Blanco, Y., and Parro, V.: Viable cyanobacteria in the deep continental subsurface, P. Natl. Acad. Sci. USA, 115, 10702–10707, https://doi.org/10.1073/pnas.1808176115, 2018.
Qin, W., Amin, S. A., Martens-Habbena, W., Walker, C. B., Urakawa, H., and Devol, A. H.: Marine ammonia-oxidizing archaeal isolates display obligate mixotrophy and wide ecotypic variation, P. Natl. Acad. Sci. USA, 111, 12504–12509, 2014.
Qin, W., Heal, K. R., Ramdasi, R., Kobelt, J. N., Martens-Habbena, W., Bertagnolli, A. D., Amin, S. A., Walker, C. B., Urakawa, H., Könneke, M., Devol, A. H., Moffett, J. W., Armbrust, E. V., Jensen, G. J., Ingalls, A. E., and Stahl, D. A.: Nitrosopumilus maritimus gen. nov., sp. nov., Nitrosopumilus cobalaminigenes sp. nov., Nitrosopumilus oxyclinae sp. nov., and Nitrosopumilus ureiphilus sp. nov., four marine ammonia-oxidizing archaea of the phylum Thaumarchaeota, Int. J. Syst. Evol. Microbiol., 67, 5067–5079, 2017.
Rieck, A., Herlemann, D. P. R., Jürgens, K., and Grossart, H.-P.: Particle-associated differ from free-living bacteria in surface waters of the Baltic Sea, Front. Microbiol., 6, 1297, https://doi.org/10.3389/fmicb.2015.01297, 2015.
Rinke, C., Rubino, F., Messer, L. F., Youssef, N., Parks, D. H., Chuvochina, M., Brown, M., Jeffries, T., Tyson, G. W., Seymour, J. R., and Hugenholtz, P.: A phylogenomic and ecological analysis of the globally abundant Marine Group II archaea (Ca. Poseidoniales ord. nov.), ISME J., 13, 663–675, 2019.
Rinta-Kanto, J. M., Sun, S., Sharma, S., Kiene, R. P., and Moran, M. A.: Bacterial community transcription patterns during a marine phytoplankton bloom, Environ. Microbiol., 14, 228–239, 2012.
Salazar, G., Cornejo-Castillo, F. M., Borrull, E., Díez-Vives, C., Lara, E., Vaqué, D., Arrieta, J. M., Duarte, C. M., Gasol, J. M., and Acinas, S. G.: Particle-association lifestyle is a phylogenetically conserved trait in bathypelagic prokaryotes, Mol. Ecol., 24, 5692–5706, 2015.
Salazar, G., Cornejo-Castillo, F. M., Benítez-Barrios, V., Fraile-Nuez, E., Álvarez-Salgado, X. A., Duarte, C. M., Gasol, J. M., and Acinas, S. G.: Global diversity and biogeography of deep-sea pelagic prokaryotes, ISME J., 10, 596–608, 2016.
Simon, M., Grossart, H. P., Schweitzer, B., and Ploug, H.: Microbial ecology of organic aggregates in aquatic ecosystems, Aquat. Microb. Ecol., 28, 175–211, 2002.
Smith, D. C., Simon, M., Alldredge, A. L., and Azam, F.: Intense hydrolytic enzyme activity on marine aggregates and implications for rapid particle dissolution, Nature, 359, 139–142, 1992.
Smith, M. W., Allen, L. Z., Allen, A. E., Herfort, L., and Simon, H. M.: Contrasting genomic properties of free-living and particle-attached microbial assemblages within a coastal ecosystem, Front. Microbiol., 4, 120, https://doi.org/10.3389/fmicb.2013.00120, 2013.
Sun, F., Wang, Y., Wu, M., Jiang, Z., Sun, C., and Cheng, H.: Genetic diversity of bacterial communities and gene transfer agents in northern South China Sea, PLoS ONE, 9, e111892, https://doi.org/10.1371/journal.pone.0111892, 2014.
Suter, E. A., Pachiadaki, M., Taylor, G. T., Astor, Y., and Edgcomb, V. P.: Free-living chemoautotrophic and particle-attached heterotrophic prokaryotes dominate microbial assemblages along a pelagic redox gradient, Environ. Microbiol., 20, 693–712, 2018.
Suzuki, S., Kaneko, R., Kodama, T., Hashihama, F., Suwa, S., Tanita, I., Furuya, K., and Hamasaki, K.: Comparison of community structures between particle-associated and free-living prokaryotes in tropical and subtropical Pacific Ocean surface waters, J. Oceanogr., 73, 383–395, 2017.
Takai, K. and Horikoshi, K.: Genetic diversity of archaea in deep-sea hydrothermal vent environments, Genetics, 152, 1285–1297, 1999.
Tarn, J., Peoples, L. M., Hardy, K., Cameron, J., and Bartlett, D. H.: Identification of free-living and particle-associated microbial communities present in hadal regions of the Mariana Trench, Front. Microbiol., 7, 665, https://doi.org/10.3389/fmicb.2016.00665, 2016.
Teeling, H., Fuchs, B. M., Becher, D., Klockow, C., Gardebrecht, A., Bennke, C. M., Kassabgy, M., Huang, S., Mann, A. J., Waldmann, J., Weber, M., Klindworth, A., Otto, A., Lange, J., Bernhardt, J., Reinsch, C., Hecker, M., Peplies, J., Bockelmann, F. D., Callies, U., Gerdts, G., Wichels, A., Wiltshire, K. H., Glöckner, F. O., Schweder, T., and Amann, R.: Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom, Science, 336, 608–611, 2012.
Tully, B. J.: Metabolic diversity within the globally abundant Marine Group II Euryarchaea offers insight into ecological patterns, Nat. Commun., 10, 271, https://doi.org/10.1038/s41467-018-07840-4, 2019.
Turley, C. M. and Mackie, P. J.: Biogeochemical significance of attached and free-living bacteria and the flux of particles in the NE Atlantic Ocean, Mar. Ecol. Prog. Ser., 115, 191–203, https://doi.org/10.3354/meps115191, 1994.
Turley, C. M. and Stutt, E. D.: Depth-related cell-specific bacterial leucine incorporation rates on particles and its biogeochemical significance in the Northwest Mediterranean, Limnol. Oceanogr., 45, 419–425, https://doi.org/10.4319/lo.2000.45.2.0419, 2000.
Varela, M. M., Vanaken, H. M., Sintes, E., and Herndl, G.: Latitudinal trends of Crenarchaeota, and bacteria, in the meso- and bathypelagic water masses of the eastern north Atlantic, Environ. Microbiol., 10, 110–124, 2008.
Wang, Y., Wang, B., Dann, L. M., Mitchell, J. G., Hu, X., Tang, H., Zhang, H., and Shen, Y.: Bacterial community structure in the Bohai Strait provides insights into organic matter niche partitioning, Cont. Shelf Res., 169, 46–54, 2018.
Wright, T. D., Vergin, K. L., Boyd, P. W., and Giovannoni, S. J.: A novel delta-subdivision proteobacterial lineage from the lower ocean surface layer, Appl. Environ. Microbiol., 63, 1441–1448, 1997.
Wu, J., Chung, S. W., Wen, L. S., Liu, K. K., Chen, Y. L. L., Chen, H. Y., and Karl, D. M.: Dissolved inorganic phosphorus, dissolved iron, and trichodesmium in the oligotrophic South China Sea, Global Biogeochem. Cy., 17, 1008, https://doi.org/10.1029/2002GB001924, 2003.
Yawata, Y., Cordero, O. X., Menolascina, F., Hehemann, J.-H., Polz, M. F., and Stocker, R.: Competition-dispersal tradeoff ecologically differentiates recently speciated marine bacterioplankton populations, P. Natl. Acad. Sci. USA, 111, 5622–5627, https://doi.org/10.1073/pnas.1318943111, 2014.
Yilmaz, P., Yarza, P., Rapp, J. Z., and Glöckner, F. O.: Expanding the world of marine bacterial and archaeal clades, Front. Microbiol., 6, 1524, https://doi.org/10.3389/fmicb.2015.01524, 2016.
Yung, C.-M., Ward, C. S., Davis, K. M., Johnson, Z. I., and Hunt, D. E.: Insensitivity of diverse and temporally variable particle-associated microbial communities to bulk seawater environmental parameters, Appl. Environ. Microbiol., 82, 3431–3437, 2016.
Zhang, R., Liu, B., Lau, S. C. K., Ki, J. S., and Qian, P.: Particle-attached and free-living bacterial communities in a contrasting marine environment: Victoria Harbor, Hong Kong, FEMS Microbiol. Ecol., 61, 496–508, 2007.
Zhang, Y., Xiao, W., and Jiao, N.: Linking biochemical properties of particles to particle-attached and free-living bacterial community structure along the particle density gradient from freshwater to open ocean, J. Geophys. Res.-Biogeo., 121, 2261–2274, https://doi.org/10.1002/2016JG003390, 2016.
Zhang, H., Wang, K., Shen, L., Chen, H., Hou, F., Zhou, X., Zhang, D., and Zhu, X.: Microbial community dynamics and assembly follow trajectories of an early-spring diatom bloom in a semienclosed bay, Appl. Environ. Microbiol., 84, e01000-18, https://doi.org/10.1128/AEM.01000-18, 2018.