the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Jun 2021
| 07 Jun 2021
Technical note: Uncovering the influence of methodological variations on the extractability of iron-bound organic carbon
Johan C. Faust
Oliver W. Moore
Caroline L. Peacock
Christian März
Association of organic carbon (OC) with reactive iron (FeR) represents an important mechanism by which OC is protected against remineralisation in soils and marine sediments. Recent studies indicate that the molecular structure of organic compounds and/or the identity of associated FeR phases exert a control on the ability of an OC–FeR complex to be extracted by the citrate–bicarbonate–dithionite (CBD) method. However, many variations of the CBD extraction are used, and these are often uncalibrated to each other, rendering comparisons of OC–FeR values extracted via the different methods impossible. Here, we created synthetic ferrihydrite samples coprecipitated with simple organic structures and subjected these to modifications of the most common CBD method. We altered some of the method parameters (reagent concentration, time of the extraction and sample preparation methods) and measured FeR recovery to determine which (if any) modifications affected the release of FeR from the synthetic sample. We provide an assessment of the reducing capacity of Na dithionite in the CBD method (the amount of Fe reduced by a fixed amount of dithionite) and find that the concentration of dithionite deployed can limit OC–FeR extractability for sediments with a high FeR content. Additionally, we show that extending the length of any CBD extraction offers no benefit in removing FeR. Moreover, we demonstrate that for synthetic OC–FeR samples dominated by ferrihydrite, freeze-drying samples can significantly reduce OC–FeR extractability; this appears to be less of an issue for natural marine sediments where natural ageing mechanisms may mimic the freeze-drying process for more stable Fe phases. While our study is not an all-inclusive method comparison and is not aimed at delivering the “perfect” extraction setup, our findings provide a collected summary of critical factors which influence the efficiency of the CBD extraction for OC–FeR. As such, we provide a platform from which OC–FeR values obtained under different methods can be interpreted and future studies of sediment carbon cycling can build upon.
- Article
(747 KB) - Full-text XML
- BibTeX
- EndNote





Marine sediments represent the largest sink for organic carbon (OC) on Earth (Hedges and Keil, 1995), and as such the preservation of OC here is crucial in controlling atmospheric carbon dioxide (CO2) levels over geological timescales (Canfield, 1993). Preservation of OC has been linked to different mechanistic and environmental factors, e.g. intrinsic recalcitrance of biomacromolecules, physical protection of OC by organic/inorganic matrices and redox conditions (Burdige, 2007, and references therein). The physical protection of OC by association with reactive iron (FeR) minerals, via mono or multi-layer adsorption and/or coprecipitation, is thought to represent a significant mechanism by which OC is preserved in marine sediments, accounting for 10 %–20 % of the sedimentary OC pool (Lalonde et al., 2012; Salvadó et al., 2015; Ma et al., 2018; Zhao et al., 2018; Wang et al., 2019; Faust et al., 2020, 2021). Additionally, the OC–FeR interaction is equally important in extending the residence time of OC in soils, for water retention (Rawls et al., 2003), resilience to erosion and overall soil fertility via nutrient bioavailability (Milne et al., 2015). The prevalence of OC–FeR is generally greater in soils than in sediments, accounting for approximately 40 % of soil total organic carbon (TOC) (Wagai and Mayer 2007; Zhao et al., 2016).
Extraction of reactive iron phases with which OC can associate has been conducted by various iterations of the citrate–bicarbonate–dithionite (CBD) method. The method was originally applied to the extraction of iron oxides from soils (Deb, 1950; Mehra and Jackson, 1958) before being adapted for OC–FeR quantification in marine sediments by Lalonde et al. (2012). The CBD extraction operates on the principle that reductive dissolution of reactive Fe phases with sodium (Na) dithionite exclusively and quantitatively liberates FeR-bound OC from the sediment matrix. This extraction can be considered to target OC–FeR since the vast majority of iron-bound OC is associated with the highly reactive (FeR) fraction, dissolved by CBD, since more crystalline Fe phases have both reduced surface reactivity (Lalonde et al., 2012) and smaller specific surface area (Jelavić et al., 2020) for OC sorption. The reductive release of OC from an OC–FeR complex has been shown to occur asynchronously, and OC is mobilised to the dissolved phase at a greater rate than Fe (Adhikari et al., 2016). The dissolution of OC–FeR is conducted at circumneutral pH buffered with sodium bicarbonate and trisodium citrate to prevent partial hydrolysis of OC (Lalonde et al., 2012). The circumneutral pH CBD extraction has also been used as part of the original SEDEX protocol for the extraction of Fe-bound phosphate (FeP) (Ruttenberg, 1992; Kraal et al., 2012). Although thermodynamically different from the CBD extraction for OC–FeR (8 h at 25 ∘C vs. 15 min at 80 ∘C), Slomp et al. (1996) found no difference between the efficiency of this phosphate extraction and the shortened high-temperature extraction of Mehra and Jackson (1958). While Ruttenberg (1992) and Thompson et al. (2019) report 90 %–100 % of synthetic ferrihydrite is extracted by the CBD method for FeP, the dithionite-to-sample ratio in their studies was more than double the ratio used in the OC–FeR extraction by Lalonde et al. (2012) (1.125 g dithionite for 0.5 g sediment vs. 0.25 g dithionite for 0.25 g sediment).
Table 1Comparison of dithionite strength to sample mass in iterations of the CBD method applied to soils and sediments.
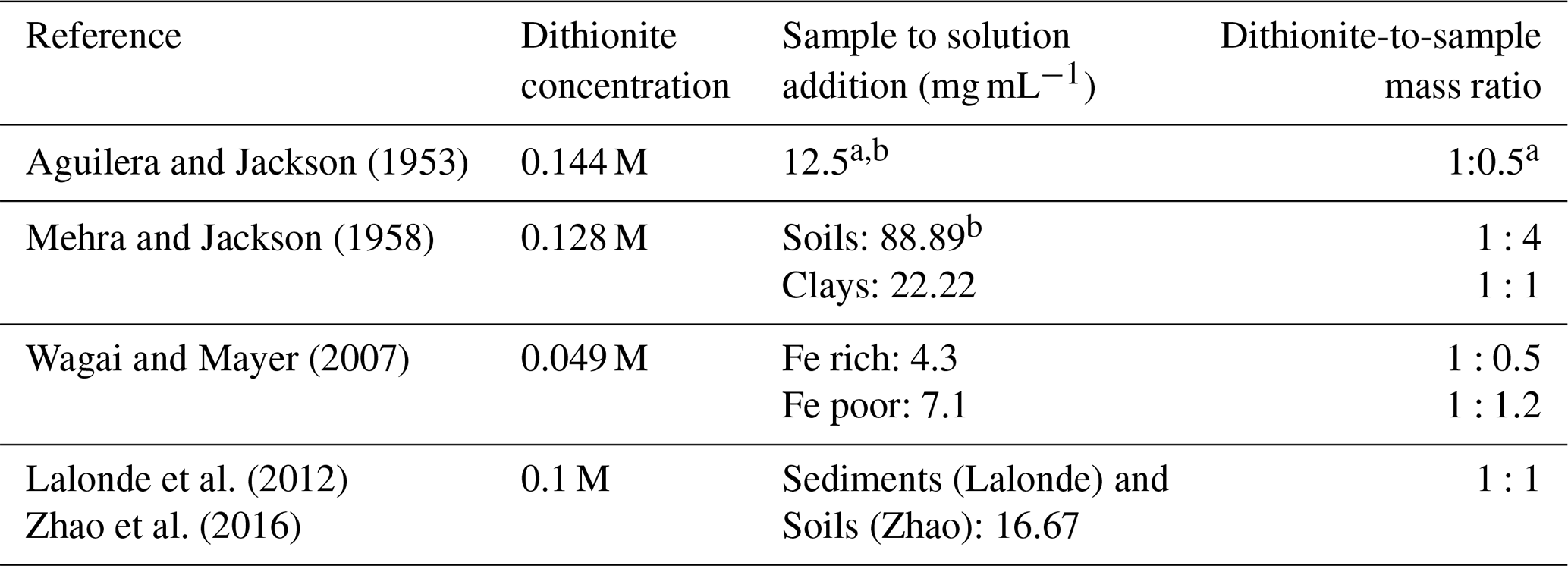
a Sample size in this method is variable due to variable Fe2O3 contents; samples should not exceed 0.5 g Fe2O3 so may be a 10 g sample with 5 % Fe2O3 content or a 1 g sample with 50 % Fe2O3 content. The ratio given is calculated on the basis of a 0.5 g Fe2O3 sample, so represents a minimal rather than absolute ratio. Dithionite concentration is based on a 40 mL reaction, while Aguilera and Jackson (1953) refer to the addition of a dithionite solution without reporting the exact volume. b If any sample exceeds 5 % Fe2O3, the extraction should be repeated an additional one to two times.
Recent findings indicate that CBD is less efficient at extracting crystalline hematite than previously thought, with 18.4 ± 0.7 % of Fe in a synthetic hematite sample recovered by Thompson et al. (2019). Similarly, this inefficiency has been shown in the context of OC–FeR extractions conducted at the lower dithionite strength where Adhikari and Yang (2015) report 5 %–44 % of OC was released from hematite–humic acid complexes upon Fe dissolution. Fisher et al. (2020) also document incomplete (< 60 %) reduction of ferrihydrite complexes by the same method (0.25 g of dithionite) and show that the molecular composition of associated OC has a large influence on Fe reactivity towards dithionite, with carboxyl-rich compounds being most resistant towards extraction. As the extraction is operationally defined based upon the susceptibility of an individual compound/mineral to chemical treatment and not upon the identity of that compound/mineral, OC composition and Fe phase crystallinity both have the ability to alter the reactivity, and therefore susceptibility, of an OC–FeR compound to extraction by CBD treatment. These findings contrast with previous understanding of the CBD method performed in an experimental context, which states that this extraction will dissolve “all solid reactive iron phases and the organic carbon associated with these phases” (Lalonde et al., 2012). Given the incomplete extraction of OC–FeR in synthetic samples, and the apparent sensitivity of the method to changes in OC composition and Fe phase, we investigated whether differences in method parameters can alter the amount of FeR associated with OC which can be extracted from a given matrix. This experiment aimed to better understand the robustness of the method and to determine how methodological variation in the CBD extraction can affect the comparability of OC–FeR extraction values. This is an important consideration to validate OC–FeR results from different studies and to build a global understanding of the extent to which OC–FeR interactions contribute to the carbon cycle.

In previous studies using the CBD method, concentrations of Na dithionite, and the ratio of Na dithionite to sample mass in the reaction, were not uniform (Table 1). Thus, the same extraction has been conducted with different “chemical strengths”, which, for an operationally defined extraction, could make comparison of results from such experiments impossible. Despite these wide variations in Na dithionite concentration, a systematic assessment of the reductive strength of dithionite for soils or sediments has not been conducted. Additionally, earlier studies make reference to repeating the extraction multiple times for Fe-rich samples (Mehra and Jackson, 1958; Aguilera and Jackson, 1953), or to altering sample mass to account for variability in Fe contents (Wagai and Mayer, 2007). Such considerations have been lost in more recent iterations of the CBD method applied to sediments (Lalonde et al., 2012), and the effect of these method alterations on OC–FeR remains largely unknown due to the lack of cross-calibration. Similarly, some studies have extended the run time of the CBD extraction from the original 15 min (Aguilera and Jackson, 1953; Mehra and Jackson, 1958; Lalonde et al., 2012). Patzner et al. (2020) performed the CBD extraction of Lalonde et al. (2012), adjusted to room temperature, over 16 h, and Wagai and Mayer (2007) also performed a 16 h extraction, adjusted to be citrate free. In this application citrate, used to complex Fe, was substituted with a weak HCl rinse to redissolve precipitated Fe in an attempt to avoid the interference of citrate in OC quantification. As the effect of extending reaction times is unknown, but appears unlikely to be of significant benefit in improving the amount of Fe liberated due to the rapid degradation of Na dithionite in aqueous form (Lister and Garvie, 1959; Lem and Wayman, 1970), we also included reaction time as a variable.
To address the question of how methodological variation affects OC–FeR extraction, due to variable dissolution of the associated FeR phase we synthesised coprecipitates of ferrihydrite with simple organic compounds and then mixed them at various ratios with marine sediment, mimicking a natural marine sediment matrix containing OC–FeR, as deployed in Fisher et al. (2020). This method allows for the creation of a synthetic marine sediment sample with a known content of reducible Fe; therefore an accurate determination of ferrihydrite extraction efficiency can be obtained within methodological error. To prepare the OC–FeR samples, we used three different organic structures with increasing carboxyl content (one COOH, two COOH or three COOH groups) to produce three different OC–FeR samples. Our approach is targeted towards testing variations in the physical parameters and chemical concentrations of the CBD method without changing the reagents used for the reductive dissolution. As such, stages of the CBD method were individually tested for different sample preparation methods (freeze-dried vs. untreated slurries), Na dithionite concentrations and extraction times. Since freeze-drying is thought to induce particle aggregation, which may artificially shield Fe phases from reduction (Chen et al., 2020), the freeze-drying method is compared with sediment slurries to determine the effect of sample preparation method on FeR extraction.
2.1 Synthesis of ferrihydrite coprecipitates
To produce synthetic OC–FeR samples two-line ferrihydrite was chosen to represent FeR as it is readily precipitated in low-temperature, oxic, circumneutral pH conditions and is therefore ubiquitous in soils and sediments (Zhang et al., 2018). In addition, it has an established ability to be experimentally coprecipitated with organic matter (e.g. Eusterhues et al., 2008, 2011, 2014).
For the OC–FeR sample using an organic structure containing two COOH groups, we coprecipitated two-line ferrihydrite with hexanedioic acid, via the method described in Fisher et al. (2020). Briefly, 3 g of hexanedioic acid (C6H10O4) was dissolved in 250 mL of deionised (DI) water with 20 g of Fe (III) nitrate nonahydrate [Fe(NO3)3 ⋅ 9H2O]. The 1 M potassium hydroxide (KOH) solution was added by titration to achieve a pH of 7.0 ± 0.3 to precipitate two-line ferrihydrite (Schwertmann and Cornell, 2000). The resultant slurry was rinsed five times in 5 L of DI water over 4 d until gravitationally settled. The pH was raised to 7 through the dropwise addition of 0.1 M NaOH solution, centrifuged (2750 g, 20 min) and the precipitate retained. The precipitate was then either immediately frozen and freeze-dried or stored at 4 ∘C for use in the sample preparation method experiment.
OC–FeR samples were also synthesised using an organic structure containing one or three COOH groups; two additional precipitates were prepared according to the method described above but substituting hexanedioic acid with pentanoic (C5H10O2) or 1,2,4-butanetricarboxylic acid (C7H10O6), respectively. The acids used therefore differ in their carboxyl group content (pentanoic-1 COOH, hexanedioic-2 COOH, 1,2,4-Butanetricarboxylic-3 COOH), a factor thought to influence their binding to FeR via bonding between carboxyl groups and mineral hydroxyls (Karlsson and Persson, 2010, 2012; Mikutta, 2011). The coprecipitations produced three OC–FeR samples with an increasing number of carboxyl groups, resulting in increasing molar ratios of 0.04 (pentanoic), 0.25 (hexanedioic) and 0.70 (1,2,4-Butanetricarboxylic). The mass of organic acids used was determined through batch coprecipitations with varying organic contents, and the masses used here represent the saturation point, where a greater addition of organic molecules did not result in an increased OC association with ferrihydrite.
2.2 Spiking of marine sediments
To spike marine sediment with the experimentally produced OC–FeR coprecipitates, different amounts of OC–FeR were added to aliquots of a marine sediment sample from the Barents Sea (sediment core depth 33.5 cm; station B6, E40; cruise JR16006; see Hopkins, 2017). This sediment was freeze-dried, ashed (650 ∘C, 12 h) to remove OC and fumigated with HCl vapour to remove inorganic carbon. The resulting material was predominantly siliciclastic in nature with a Fe content of 16.33 mg/g. The ratio of OC–FeR coprecipitate relative to marine sediment was differed to create a content matrix; e.g. the sample referred to as “20 % OC–FeR” with a total mass of 0.25 g contains 0.200 g (80 wt %) of marine sediment and 0.50 g (20 wt %) of the OC–FeR coprecipitate. The full composition of all samples is detailed in Table 2. Mixing was achieved by agitation of either the freeze-dried coprecipitate with the sediment or the dry-weight equivalent of untreated slurry samples with the sediment. Dry weight of slurry samples was determined by drying 10×1 mL aliquots of coprecipitate slurry at 40 ∘C to calculate milligrams per millilitre of coprecipitate and taking the mean value.
2.3 Citrate–bicarbonate–dithionite reduction of FeR
To reduce FeR in the synthetic freeze-dried and untreated slurry OC–FeR coprecipitates that were spiked into the marine sediment, reductive dissolution of reactive Fe phases was conducted according to an established CBD protocol (Lalonde et al., 2012; Salvadó et al., 2015). The synthetic sediment sample (0.25 g, or dry-weight equivalent for slurry samples) was added to 13 mL of 0.11 M sodium bicarbonate (NaHCO3) and 0.27 M trisodium citrate (Na3C6H5O7) solution in a 50 mL centrifuge tube and then placed in a water bath at 80∘C to pre-heat. Subsequently, 0.25 g of sodium dithionite was dissolved in 2 mL of 0.11 M NaHCO3 and 0.27 M Na3C6H5O7 solution and added to the pre-heated mixture before vortexing and further heating at 80 ∘C for 15 min. A parallel control extraction was conducted, replacing Na dithionite and trisodium citrate with Na chloride at an equivalent ionic strength: 13 mL of 1.6 M NaCl and 0.11 M NaHCO3, followed by 0.22 g NaCl dissolved in 2 mL of the 1.6 M NaCl and 0.11 M NaHCO3 solution. Following the extraction, samples were centrifuged (3000 g, 10 min) and the supernatant was retained. A three-time rinse cycle using artificial seawater was then conducted on the precipitate to remove any residual dissolved Fe; a 15 mL aliquot from each of these rinses was retained and combined per sample. All supernatants were acidified to pH < 2 with 12 N HCl to prevent Fe precipitation.
2.4 Testing the impact of methodological variations
To test the impact of different sample preparation methods, the results from the extraction of the synthetic freeze-dried samples and the untreated slurry samples from the spiked marine sediment aliquots were compared. To test the impact of different extraction conditions, the amount of Na dithionite added to a reaction was varied, using amounts both lower (0.125 g) and higher (0.375, 0.500, 0.625 g) than the standard addition of 0.25 g Na dithionite per 0.25 g of dried sample. Where the dithionite addition was changed in the reduction reaction, an equivalent change was made for the control experiment to maintain the equivalence of ionic strengths. To test the impact of different reaction times, this was increased to 30, 45 and 60 min with manual shaking of each sample every 15 min. For the sample preparation and Na dithionite experiments, repeats of the same coprecipitate were conducted over the content matrix, differing by the amount of OC–FeR added to the sediment. While performing these extractions on pure synthetic OC–FeR is useful for uncovering a mechanistic trend, dilution with carbon-free sediment to lower OC–FeR contents in the sample ensured any trends uncovered are noticeable at environmentally relevant conditions (OC–FeR < 50 wt %). Repeats of samples across this content gradient are in lieu of direct replicates for each unique sample condition. These were not possible due to yield limitations imposed by ferrihydrite coprecipitate synthesis (net ∼ 5 g per 5 L rinse solution). All samples within any one experiment originated from the same batch of ferrihydrite.
2.5 Environmental sample treatment
To allow comparison between sample preparation methods applied to samples containing synthetic OC–FeR, natural samples were subject to the same methods of CBD extraction and Fe elemental analysis. Arctic Ocean seafloor sediment was collected (cruise: JR17007, station B16, sediment depth 22–23 cm; Faust et al., 2021), of which half was thawed following freezing at the point of sampling, and half was freeze-dried.
2.6 Elemental analysis for iron
Initial Fe contents in synthetic samples were obtained by digesting ∼ 2 mg of dried sample in 1 mL 12 N HCl at room temperature followed by a 10-fold dilution of the extract with 1 % HCl solution. Dilutions of initial samples, in addition to the extraction supernatants, were conducted using Milli-Q water to produce a subsample within the 1–10 ppm Fe range. Iron contents for both the initial samples and supernatants from the extraction were determined by atomic absorption spectroscopy (Thermo Fisher iCE3300 AAS). Calibration was performed using matrix-matched standards, and quality control was confirmed following every 10 samples by repeat sampling of calibration standards to check for drift. Supernatants from control experiments were also measured for Fe content, and these were diluted 20-fold to prevent salt blockages; the supernatant from seawater rinses remained undiluted except for where the Fe concentration in solution was > 10 ppm, whereby these were diluted 10-fold. The recovery of Fe following the extractions was calculated by subtracting the control-corrected loss of Fe from the initial sample Fe content. Maximal extraction of Fe is defined as the point from which further addition of Na dithionite does not further increase the extraction of Fe.
2.7 Elemental analysis for carbon
Carbon content was determined for all OC–FeR contents (20 %–50 %) both before and after Fe extraction to determine whether OC–FeR was completely recovered (given that the Fe this OC is bound to was incompletely reduced across the series). Carbon contents were measured using a LECO-SC144DR C&S analyser. The LECO analyser was calibrated with, and quality control checked against, a known standard (LECO 502-694). All carbon samples were analysed in an oven-dried state (40 ∘C, 12 h). Carbon recovery was calculated by subtracting the remaining carbon content from the solid phase of the extracted sample from the initial carbon content. Correction by normalisation to sample mass was made to account for mass loss during the dissolution. Instrument error for the LECO analyser was low (≤ 1 % RSD) due to drift calibration throughout the analytical run.
3.1 The effect of varying addition of Na dithionite on Fe extractability
The CBD method of Lalonde et al. (2012) requires a 0.25 g addition of Na dithionite per 0.25 g of dried sediment sample; here we adjusted the mass of dithionite added (0.125, 0.375, 0.500, 0.625 g), and the % Fe extracted from these samples was measured (Fig. 1). This can be interpreted as the reduction capacity of Na dithionite relative to initial Fe content. Our results showed that all samples have incomplete reduction of Fe, regardless of Na dithionite addition, with the highest recovery of Fe in samples with low OC–FeR contents. The 30 wt % and 40 wt % OC–FeR-containing samples show almost identical trends for their Fe extractability, while the 20 wt % OC–FeR sample was more readily extracted and the 50 wt % OC–FeR sample was least extracted.
For sediment samples containing 20 wt % OC–FeR, maximal Fe extraction occurs at the original 0.25 g dithionite addition (89 %), while maximal Fe extraction occurs with greater dithionite additions for samples with a greater initial OC–FeR content. For the 30 wt % and 40 wt % OC–FeR samples, maximal Fe extraction (∼ 88 %) occurs at 0.5 g Na dithionite addition. For the 50 wt % OC–FeR sample, 60 % of total Fe was extracted at both 0.5 and 0.625 g Na dithionite additions. Thus, the maximal % Fe in sediment extractable by 0.25 g Na dithionite lay between a 20 wt % and 30 wt % OC–FeR composition, equivalent to 7 wt %–10 wt % FeR content in the sediment. Therefore, assuming a 0.25 g sample size, the absolute amount of Fe which could be extracted for any sample would be between 17.5 and 25 mg. OC–FeR extracted (i.e. OC recovery) was determined at the point at which maximal Fe extraction was achieved by addition of excess Na dithionite. The OC–FeR results (Fig. 1) indicate OC–FeR extraction to be incomplete (< 100 %) in all our experiments.
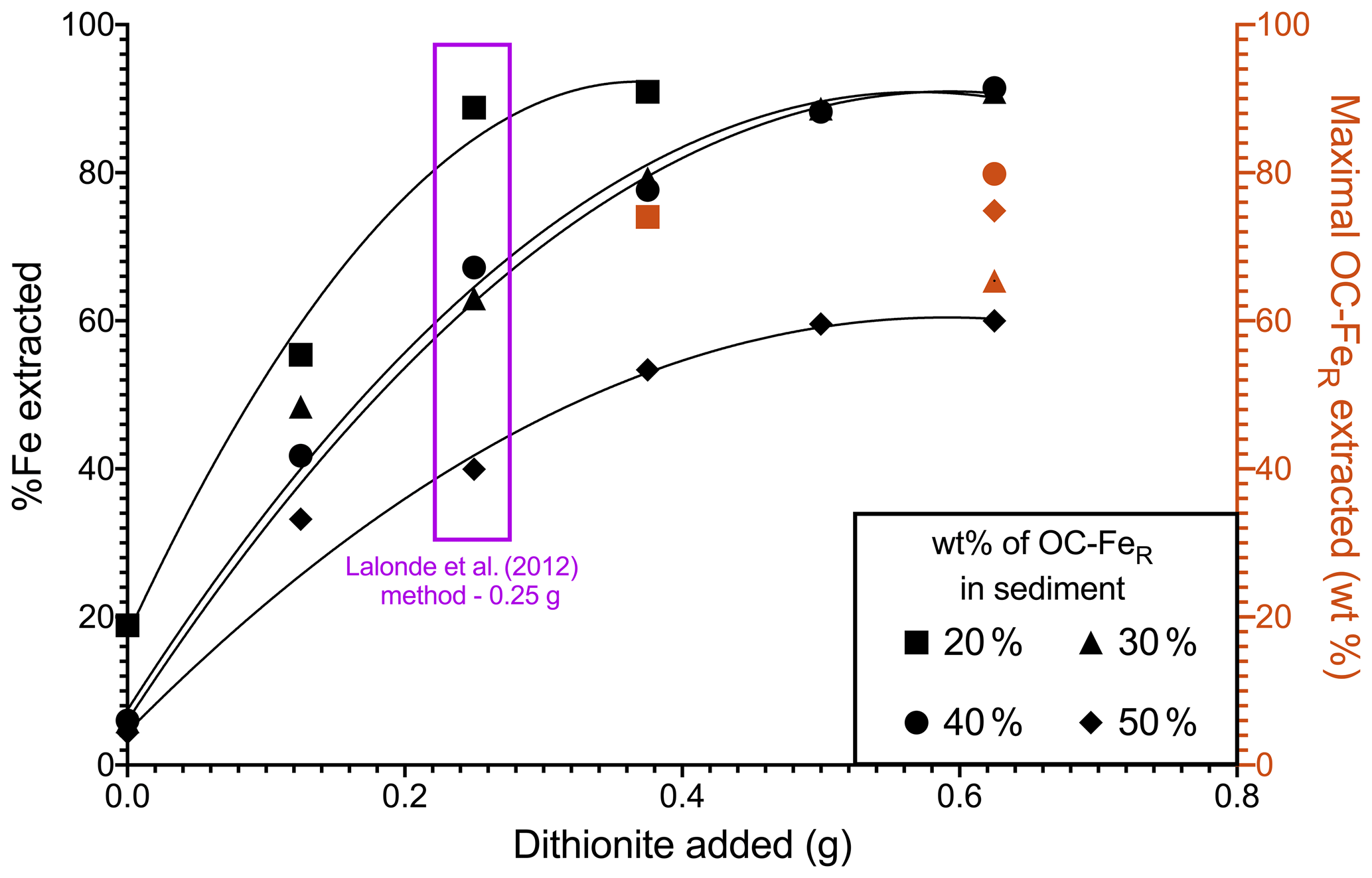
Figure 1Reduction capacity of Na dithionite in the extraction estimated from % Fe extracted with varying Na dithionite additions across an OC–FeR concentration gradient. Red shapes indicate the amount of OC–FeR extracted for the concentration of Na dithionite at which Fe is maximally extracted for that sample (black). Compound maximal instrument error is minimal and can be found in the data asset.
3.2 The effect of sample preparation methods on Fe extractability
A greater proportion of Fe is extracted from the marine sediment spiked with slurry samples (referred to as “wet”) than from the same sediments spiked with freeze-dried samples (referred to as “dry”) for all coprecipitates at all OC–FeR contents (Fig. 2). The dry spiked sediment samples achieved a maximum Fe extractability of 71 % (for the sediment spiked with 3 COOH OC–FeR, at 60 wt % OC–FeR content), while for the wet spiked sediment samples up to 87 % is recovered (with 3 COOH OC–FeR, at 100 wt % OC–FeR content). A 100 % recovery of added Fe was not achieved in any of the experiments.
Iron extractability increases with the number of carboxyl groups in the OC–FeR coprecipitate for both dry and wet spiked sediment samples; this trend is clearly shown for sediments with 100 wt %, 60 wt % and 40 wt % OC–FeR content. However, the 1 COOH OC–FeR-spiked sample at the 80 wt % OC–FeR ratio was extractable for a greater mean amount of Fe compared to the 2 COOH OC–FeR-spiked sample for both the dry and wet preparation methods. Additionally, no trend in Fe extractability with the number of carboxyl groups was present for the 20 wt % OC–FeR spiked sediment samples; however, Fe contents at this level are comparatively low, which may obscure trends within this data series.
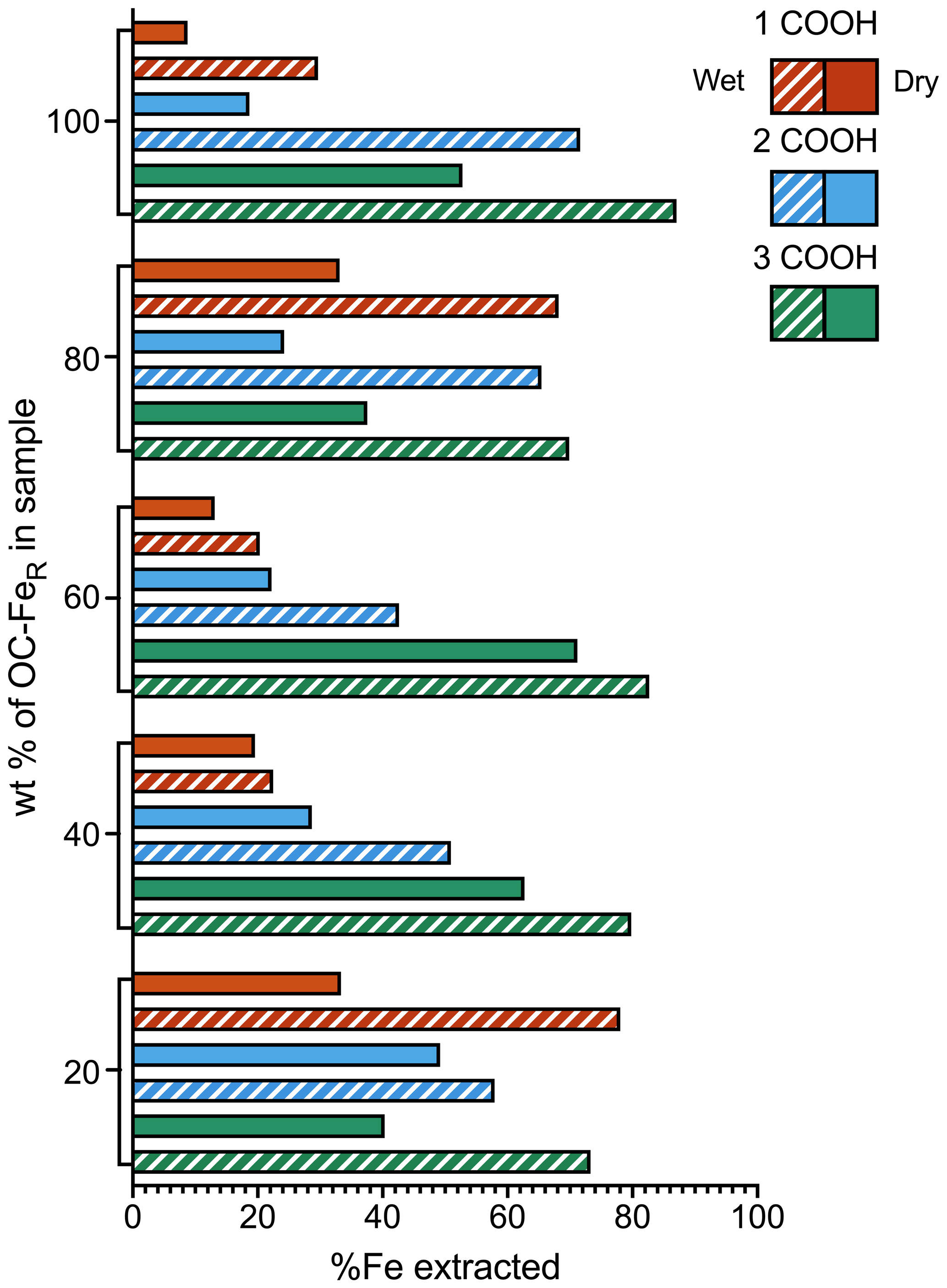
Figure 2Fe recovery from freeze-dried vs. slurry coprecipitates. Solid bars show dried samples while patterns show the wet (slurry) samples. 1, 2 and 3 COOH refer to the number of carboxyl groups present in the coprecipitated organic acids. Compound maximal instrument error is minimal and can be found in the data asset.
Following the experiments with marine sediment spiked with synthetic OC–FeR compounds, a similar investigation was conducted on marine sediment samples where no OC–FeR had been added. This experiment only compared freeze-dried vs. freeze-thawed (not slurry) samples. There was no notable difference in the amount of Fe extracted for sediment samples which had been freeze-dried compared to those which were freeze-thawed. Recovery of Fe from freeze-dried samples was 22.34 % Fe (± 4.05 (1 SD)) compared to 22.68 % (± 6.67) for freeze-thawed samples.
3.3 The effect of CBD extraction time on Fe extractability
Here we examined the effect of longer (> 15 min) extraction time periods on the Fe extraction. All other parameters of the extraction remained the same as in Lalonde et al. (2012). A 2 COOH OC–FeR spiked sediment sample at a 60 wt % OC–FeR content was used in dry and wet forms. Extraction time was extended in 15 min increments from 15 to 60 min, and results are shown in Fig. 3. The percentage of Fe extracted remained very similar across the time series for both wet and dry samples, and there was no evidence that increasing the extraction duration systematically increases Fe liberation.

Figure 3% Fe extracted across a time series for CBD extraction. The sample used in this experiment was a spiked sediment comprised of 60 wt % sediment and 40 wt % of a 2 COOH OC–FeR coprecipitate. Compound maximal instrument error is minimal and can be found in the data asset.
4.1 Concentration of Na dithionite as a primary control on OC–FeR extraction
Here, we altered the CBD method (Lalonde et al., 2012) by changing the mass of Na dithionite added to the CBD extraction for four synthetic sediments, each differing in Fe content, to determine whether an increased concentration of Na dithionite affects Fe liberation.
The four synthetic samples subjected to dithionite reduction (20 wt %–50 wt % initial OC–FeR content) differed in Fe content (7 wt % Fe–24 wt % Fe), resulting in a dithionite-to-Fe mass ratio of 1:0.07–0.24. These ratios represent a stronger reductive power than the concentration of dithionite previously used in Fe extractions, where the chosen method resulted in < 100 % extraction of the targeted phases. For example, Adhikari and Yang (2015) report < 50 % Fe was extracted with a dithionite-to-Fe ratio of 1:0.8 for a humic-hematite complex. Our high Fe content is driven by low ratios since only short-chain organic compounds are associated with the FeR phases and are therefore designed to be a mechanistic model rather than to simulate the types of compounds which occur naturally. Both the amount of reactive Fe and the amount of OC associated with Fe are highly variable, and the factors which control the OC–FeR interaction remain poorly understood. Reactive Fe content in many marine sediments is below 7 wt % Fe (e.g. Canfield, 1989; Raiswell and Canfield, 1998); however, spatial and temporal variation in Fe fluxes to the seafloor can result in unusually Fe-rich sediments, e.g. near hydrothermal vents (Poulton and Canfield, 2006) or in Fe–Mn nodules (Hein et al., 1997). Additionally, OC–FeR has a typical value of between 10 %–20 % (of total OC) (Lalonde et al., 2012; Salvadó et al., 2015; Ma et al., 2018; Zhao et al., 2018; Wang et al., 2019; Faust et al., 2020, 2021) yet has been observed at contents exceeding 40 % in terrestrial environments (Zhao et al., 2016; Patzner et al., 2020) and 50 % in sandy beach sediments of subterranean estuaries (e.g. 56.31 % ± 5.56 Martinique Beach, Canada; Sirois et al., 2018). This large variability in environments containing OC–FeR, and the composition of such compounds, highlights the need to examine their extractability across a wide matrix. We find that the sample containing 20 wt % OC–FeR (∼ 7 wt % Fe) is maximally extracted for its reactive Fe component under the 0.25 g (0.1 M) treatment (Fig. 1). Maximal extraction is defined as the point at which further additions of dithionite do not increase Fe liberation. Here, the sample containing 20 wt % OC–FeR liberates 88.79 % ± 3.55 of FeTotal with a 0.25 g dithionite addition, and increasing this to 0.375 g of dithionite only marginally increases Fe liberation to 90.94 % ± 3.64; as these values are within error of each other, we conclude that maximal extraction is achieved with the lower 0.25 g dithionite content.
At increased OC–FeR contents, 0.25 g of Na dithionite seems to be no longer sufficient to achieve maximal extraction. The samples with 30 wt % and 40 wt % OC–FeR content, which follow almost identical trajectories (Fig. 1), reach maximal extraction at 0.5 g 0.2 M with 88.65 % ± 3.54 and 88.22 % ± 3.53 of FeTotal recovered, respectively. These values are within the error of maximal extraction for 20 wt % OC–FeR and are significantly higher than the amount of Fe liberated under the standard 0.25 g 0.1 M extraction (63.03 % ± 2.52 and 67.21 % ± 2.69, respectively). This finding demonstrates that in OC–FeR-rich sediments, the 0.1 M or weaker Na dithionite methods could underestimate the true OC–FeR content of these samples. In any case, comparisons of OC–FeR extracted by different dithionite contents < 0.2 M for these sediments would not be comparable due to underestimations in weaker treatments. While these high-OC–FeR-content sediments of 30 %–40 % OC–FeR (of total OC) are above the average for natural marine sediments, they do exist in areas with high OC accumulation rates (e.g. shallower waters on coastal shelves) where ∼ 45 % of global OC burial is thought to occur (Hedges and Keil, 1995), and sediments have been documented as exceeding 30 % OC–FeR (e.g. equatorial Pacific 0∘ N, 34.79 %; Barber et al., 2017).
The indication that Na dithionite at the 0.25 g 0.1 M addition is increasingly inefficient with increasing OC–FeR content is reinforced at the 50 wt % OC–FeR (24 wt % Fe) composition. The amount of Fe extracted is increased from 39.96 % ± 1.60 with 0.1 M (0.25 g) Na dithionite to 59.58 % ± 2.38 at double strength (0.2 M). This differs from the previous compositions in reaching a maximum at ∼ 60 % Fe, as opposed to the ∼ 90 % achieved for 20 wt %–40 wt % OC–FeR. Given that % Fe removed does not increase with further addition of Na dithionite (0.625 g), the amount of Na dithionite is no longer the limiting factor in extracting Fe from such very OC–FeR-rich samples (Fig. 1). It might be that another reagent, potentially trisodium citrate, becomes limiting. In the extraction reaction, citrate acts as a complexing agent to keep Fe dissolved in solution (Lalonde et al., 2012; Sirois et al., 2018). If the increased strength dithionite treatment increases dissolved Fe beyond the complexing capacity of citrate, then excess Fe likely precipitates out of solution before measurement. However, Henkel et al. (2016) found that a reduced concentration of citrate is sufficient to fully complex the reduced Fe pool when the extraction is performed under anoxic conditions, which may remove the need to further increase the addition of citrate as an organic reagent.
Excess Fe precipitation out of solution may further explain the observation in OC extraction for the 50 wt % OC–FeR sample whereby this sample appears to experience a greater loss of OC compared to Fe during the extraction, despite a molar ratio of < 1. In reality, it is likely that the actual FeR loss is greater than detected for this sample, due to Fe precipitation before measurement, and greater than the measured OC loss, following the expected trend based on the ratio. This is observed for all other samples with a lower initial OC–FeR content where FeR loss was not thought to be influenced by excess precipitation and FeR loss was greater than that for OC (Fig. 1). Nevertheless, our results show that out of the varied experimental parameters, increasing the amount of Na dithionite in the reaction had the greatest effect in increasing Fe liberation for samples containing > 20 wt % OC–FeR (∼ 7 wt % Fe). Therefore, this indicates that for OC–FeR-rich sediments (≥ 30 wt %), OC–FeR extraction values determined by methods which differ in their concentrations of dithionite below 0.2 M are not comparable, since for these samples OC–FeR continues to be extracted with increasing additions of Na dithionite.
4.2 Sediment freeze-drying as a limiting factor on Fe reduction
Chemical extractions of Fe are typically performed on freeze-dried sediment samples. Removal of the aqueous phase decreases sample mass and prevents the need for frozen storage, and the biochemical profile is preserved through inhibiting microbial degradation. Alternative treatments such as air drying are considered to be more aggressive as they can alter the chemical composition of samples and may inflict significant changes on sediment chemistry, including losses of biomarkers (McClymont et al., 2007) and changes in speciation of heavy metals (Zhang et al., 2001). However, how this process affects the physical properties of samples and their subsequent behaviour towards chemical reagents has not been widely considered. We found that the Fe extraction efficiency from freeze-dried sediment samples containing freeze-dried freshly precipitated ferrihydrite was much lower than that measured for chemically identical samples containing freshly precipitated ferrihydrite present in slurry form, i.e. not freeze-dried (Fig. 2).
In line with previous studies, we suggest that freeze-drying may result in reduced Fe extractability compared to not freeze-dried samples due to particle aggregation or transformation of ferrihydrite to a more stable phase. Aggregation can produce “shielded” sediment particles (Chen et al., 2020), and this may inhibit Fe reduction by reducing the reactive mineral surface area exposed to dithionite. This could be overcome, e.g. by crushing, but would introduce further variability, e.g. in grain size (Raiswell et al., 1994), and would be unlikely to be effective against nanoparticles. The influence of freeze-drying on particle size has been previously noted, particularly for sediment with a high clay content (> 39 %) (Keiser et al., 2014). McKeague and Day (1966) similarly report that finer grinding of sediment resulted in an increased extraction of Fe. These findings indicate that particle size is a critical parameter in determining the amount of Fe extracted; however, current methods do not define the particle size of “finely ground” sediments. An alternate hypothesis, that mineralogical transformation of ferrihdyrite during freeze-drying may lead to reduced Fe recovery, was ruled out by X-ray diffraction (XRD) characterisation of a representative freeze-dried sample, which confirmed the identity to still be two-line ferrihydrite (Fisher et al., 2020).
To reduce the effect of aggregation during freeze-drying, a few studies on soils used fresh slurry samples (e.g. van Bodegom et al., 2003; Chen et al., 2020). Wet thawed samples have been used more widely in the sequential extraction of Fe (e.g. Wehrmann et al., 2014; Riedinger et al., 2017; Laufer et al., 2020), and the Arctic marine sediment sample used in our analysis was similarly thawed following freezing on collection. We find that our thawed sample shows no difference in its recovery for Fe compared to the dried variant of this sample. Natural ageing processes within the sediment could explain this lack of freeze-drying effect in older sediments due to both the physical effects of ageing on Fe minerals and the transformation of poorly crystalline reactive ferrihydrite phases to more crystalline phases at 22 cm depth (Faust et al., 2021), which are unlikely to be extracted by the neutral pH CBD extraction.
Although our results show that extraction efficiency of Fe from ferrihydrite is better in wet sediment samples, it is still only possible to achieve a maximal extraction efficiency of 87 % (with 3 COOH OC–FeR, at 100 wt % OC–FeR content) (Fig. 2). The extraction efficiency for FeR phases associated with less complex OC is even lower, e.g. only 30 % of Fe is liberated for the 1 COOH OC–FeR-spiked sediment sample at the same OC–FeR content (100 wt %). Even though such samples consisting exclusively of ferrihydrite-associated OC are unlikely to occur in nature, on the whole our results confirm previously identified relationships between Fe extraction and carboxyl content, where Fe extraction using the CBD method is more efficient for OC–FeR that contains more carboxyl-rich OC (Fisher et al., 2020). This is attributed to the greater amorphicity of ferrihydrite coprecipitated with carboxyl-rich OC; i.e. the resultant mineral phase is less crystalline than ferrihydrite coprecipitated with less-carboxyl-rich OC and is therefore easier to reductively dissolve. An inflation of the Fe extractability for the 1 and 2 COOH OC–FeR-spiked sediment samples at the 20 wt % OC–FeR content is likely due to uncertainty as a result of the dilution. The 1 COOH OC–FeR-spiked sediment sample at 80 wt % OC–FeR also appears inflated and out of step with the trends set by the samples at other OC–FeR contents, with no obvious explanation.
While application of the CBD method to slurried samples could increase the extracted proportion of Fe associated with OC, such an approach may not always be practical, either due to practical considerations, such as the difficulty in transporting heavy wet sediments, or when there is a need to preserve the sediment profile, for example, protecting anoxic sediments from oxic biological transformations. Additionally, the inability to fully extract Fe even when sediments are in a slurried state indicates that other limiting factors to Fe extractability persist, which prevent complete extraction of FeR phases by the circumneutral CBD method. Nevertheless, our results confirm that the process of sample preparation has a large effect on Fe recovery, with non-freeze-dried synthetic OC–FeR samples that contain ferrihydrite and carboxyl OC being extractable for a much greater proportion of Fe compared to their freeze-dried equivalents. However, this finding was not replicated for naturally aged Arctic Ocean samples, where FeR content was dominated by more stable phases such as hematite and goethite, indicating that older natural sediments are less influenced by freeze-drying compared to freshly precipitated ferrihydrite.
4.3 Rapid reduction of FeR by Na dithionite
One parameter of the extraction method which has remained largely consistent across all iterations of CBD treatment is the extraction length of 15 min (Mehra and Jackson, 1958; Wagai and Mayer, 2007; Lalonde et al., 2012). As we observed incomplete Fe extraction (Fig. 1) for all our synthetic samples, a range of CBD extraction times were trialled to understand whether reaction time and Fe extraction have a positive correlation, as seen in some iterations of the CBD method for OC–FeR (e.g. Wagai and Mayer, 2007). In some applications of the CBD method, the extraction stage is repeated multiple times for the same sample in order to fully extract FeCBD (e.g. Aguilera and Jackson, 1953; Mehra and Jackson, 1958), but it is still difficult to attribute full CBD extraction to this multiple extraction protocol because the parameter that prevents full extraction of CBD-extractable Fe in the first place is unknown.
Exposure time to CBD of freeze-dried and slurried OC–FeR synthetic samples spiked into marine sediment (using 2 COOH, OC–FeR at 40 wt % OC–FeR content) was increased from a 15 min treatment in 15 min intervals up to 60 min total exposure time (Fig. 3). No difference was observed for the amount of extractable Fe across the time series, and we therefore propose that an increase in chemical exposure time has no effect on Fe extractability for OC–FeR phases in natural samples, and reductive dissolution of the susceptible Fe phases occurs relatively fast. This finding is in agreement with the fact that dithionite, the reductive component, is known to undergo degradation to form sodium thiosulfate and bisulfite in aqueous solutions with a rapid second-order rate constant (K2) of 3.0 mol L−1 min−1 at 79.4 ∘C; hence reducing conditions are unlikely to be sustained for long (Lister and Garvie, 1959). However, Patzner et al. (2020) adapted the CBD method whereby time was extended to compensate for a reduction in the temperature of the reaction. A low-temperature approach was not tested in our study as we focused on thermodynamically increasing the efficiency of the reaction; however, this adaptation may prove useful should non-destructive analysis be required. For example, subsequent analysis of biomarkers in extracted organics is currently not possible due to temperature-induced transformation and degradation of OC when heated to 80 ∘C. These types of analyses may allow us to better understand the origins and molecular composition of organic matter (OM) involved in mineral-based preservation processes and offers promising scope for future experimentation with the CBD method.
Reductive dissolution of OC–FeR using the CBD method is an important and widely used protocol for quantifying mineral-associated OC in sediments and soils. In this study we aimed to address the uncertainty around variations in the preparation method for samples subject to CBD and some of the method parameters used during the extraction, to understand whether these factors have an effect on the extraction efficiency of OC–FeR and therefore the interpretation of OC–FeR data. Our results show that the mass of dithionite added to a sample has a strong control on the extractability of the easily reducible Fe pool and that this is particularly acute for reactive Fe-rich sediments, where a doubling of Na dithionite addition for these sediments can increase FeR recovery from ∼ 60 % to ∼ 90 %. While a 0.1 M Na dithionite concentration appears to be sufficient for most marine sediments with average reactive Fe contents, data produced with lower dithionite-to-solid ratios should not be compared to those extracted by a greater concentration of dithionite. Earlier studies, e.g. Mehra and Jackson (1958) and Wagai and Mayer (2007), accounted for this issue by varying the dithionite-to-solid ratio based on Fe contents. We suggest that this approach should be redeployed for the extraction of FeR from Fe-rich environments (> 30 % OC–FeR), whereby an increase in dithionite concentration was shown to extract a greater amount of OC–FeR than the standard 0.1 M approach. We also show that freeze-drying reduces Fe liberation from synthetic coprecipitates containing ferrihydrite when compared to the equivalent sample in slurried form, likely due to particle aggregation. However, the effects of freeze-drying are negligible for aged sediments where the reactive Fe mineralogy is dominated by more stable phases. While we recognise that the use of fresh sediment slurries is rarely practical, we suggest that for all samples, particularly the uppermost surface sediments, grain size or the grinding method should be reported alongside OC–FeR extractions to aid comparability of samples beyond current descriptions of “finely ground”. Where sediment slurries are used, 10 aliquots should be oven-dried to determine the dry mass equivalent of the sample to ensure the dithionite-to-solid ratio is maintained; for our samples we find that variability in the dry mass contained in a slurry sample introduces an error of up to ± 5 %. Finally, we show that an increase in reaction time (up to 1 h) results in no additional Fe extractability over the typical 15 min reaction time. Given the variability in often uncalibrated extraction protocols within the literature, we show that comparison of the results from different studies is problematic when attempting to elucidate the true extent of OC–FeR in the global carbon cycle. Future work to quantify the global importance of the OC–FeR sink requires a uniform methodological approach to be deployed across a range of environments, with modification for FeR-rich environments. These extractions should be conducted with the knowledge that this represents the operationally defined, not absolute, OC–FeR content of sediments.
The equation used to compute OC–FeR is available in Fisher et al. (2020); examples of implementation can be found in the associated electronic annex of that paper (https://data.mendeley.com/datasets/gpt8f8kpcs/1, last access: 17 May 2021). Raw figure data for this paper are available via the defined asset at https://doi.org/10.6084/m9.figshare.14384276 (last access: 17 May 2021, Fischer et al., 2021).
BJF, JCF, OWM, CLP and CM designed the study, and BJF conducted the experimental work with support from JCF and OWM. JCF supplied the Arctic sample material. BJF, JCF, OWM, CLP and CM all contributed towards analysis of the data sets. BJF prepared the first draft of the manuscript, and all authors contributed towards review and editing of the final version.
The authors declare that they have no conflict of interest.
This work was supported by funds from the ChAOS project (NE/P006493/1), part of the Changing Arctic Ocean programme, jointly funded by the UKRI Natural Environment Research Council (NERC) and the German Federal Ministry of Education and Research (BMBF). Additionally, this research project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 725613 MinOrg). Caroline L. Peacock gratefully acknowledges the Royal Society Wolfson Research Merit Award (WRM/FT/170005). We thank Andrew Hobson and Fiona Keay of Cohen Geochemistry, University of Leeds for their assistance with sample analysis. We are grateful for the detailed and insightful reviews by Susann Henkel, Peter Kraal, Tom Jilbert, Jeffery Cornwell and the two anonymous reviewers which have significantly improved the manuscript, as well as Jack Middelburg for editorial handling.
This research has been jointly supported by the Natural Environment Research Council and Bundesministerium für Bildung und Forschung (grant no. NE/P006493/1). This research was also supported by the European Research Council, H2020 European Research Council (grant no. MINORG (725613)) and a Natural Environment Research Council Highlight Topic Grant (NE/S004963/1 Locked Up).
This paper was edited by Jack Middelburg and reviewed by Susann Henkel, Tom Jilbert, Peter Kraal, Jeffrey C. Cornwell, and two anonymous referees.
Adhikari, D. and Yang, Y.: Selective stabilization of aliphatic organic carbon by iron oxide, Sci. Rep., 5, 11214, https://doi.org/10.1038/srep11214, 2015.
Adhikari, D., Poulson, S. R., Sumaila, S., Dynes, J. J., McBeth, J. M., and Yang, Y.: Asynchronous reductive release of iron and organic carbon from hematite–humic acid complexes, Chem. Geol., 430, 13-20, https://doi.org/10.1016/j.chemgeo.2016.03.013, 2016.
Aguilera, N. H. and Jackson, M. L.: Iron Oxide Removal from Soils and Clays, Soil Sci. Soc. Am. J., 17, 359–364, https://doi.org/10.2136/sssaj1953.03615995001700040015x, 1953.
Barber, A., Brandes, J., Leri, A., Lalonde, K., Balind, K., Wirick, S., Wang, J., and Gelinas, Y.: Preservation of organic matter in marine sediments by inner-sphere interactions with reactive iron, Sci. Rep., 7, 366, https://doi.org/10.1038/s41598-017-00494-0, 2017.
Canfield, D. E.: Reactive iron in marine sediments, Geochim. Cosmochim. Ac., 53, 619–632, https://doi.org/10.1016/0016-7037(89)90005-7, 1989.
Chen, C., Hall, S. J., Coward, E., and Thompson, A.: Iron-mediated organic matter decomposition in humid soils can counteract protection, Nat. Commun., 11, 2255, https://doi.org/10.1038/s41467-020-16071-5, 2020.
Eusterhues, K., Wagner, F. E., Hausler, W., Hanzlik, M., Knicker, H., Totsche, K. U., Kogel-Knabner, I., and Schwertmann, U.: Characterization of ferrihydrite-soil organic matter coprecipitates by X-ray diffraction and Mossbauer spectroscopy, Environ. Sci. Technol., 42, 7891–7897, https://doi.org/10.1021/es800881w, 2008.
Eusterhues, K., Rennert, T., Knicker, H., Kogel-Knabner, I., Totsche, K. U., and Schwertmann, U.: Fractionation of organic matter due to reaction with ferrihydrite: coprecipitation versus adsorption, Environ. Sci. Technol., 45, 527–533, https://doi.org/10.1021/es1023898, 2011.
Eusterhues, K., Hädrich, A., Neidhardt, J., Küsel, K., Keller, T. F., Jandt, K. D., and Totsche, K. U.: Reduction of ferrihydrite with adsorbed and coprecipitated organic matter: microbial reduction by Geobacter bremensis vs. abiotic reduction by Na-dithionite, Biogeosciences, 11, 4953–4966, https://doi.org/10.5194/bg-11-4953-2014, 2014.
Faust, J. C., Stevenson, M. A., Abbott, G. D., Knies, J., Tessin, A., Mannion, I., Ford, A., Hilton, R., Peakall, J., and März, C.: Does Arctic warming reduce preservation of organic matter in Barents Sea sediments?, Philos. Trans. Roy. Soc. A, 378, 20190364, https://doi.org/10.1098/rsta.2019.0364, 2020.
Faust, J. C., Tessin, A., Fisher, B. J., Zindorf, M., Papadaki, S., Hendry, K. R., Doyle, K. A., and März, C.: Millennial scale persistence of organic carbon bound to iron in Arctic marine sediments, Nat. Commun., 12, 275, https://doi.org/10.1038/s41467-020-20550-0, 2021.
Fisher, B. J., Moore, O. W., Faust, J. C., Peacock, C. L., and März, C.: Experimental evaluation of the extractability of iron bound organic carbon in sediments as a function of carboxyl content, Chem. Geol., 556, 119853, https://doi.org/10.1016/j.chemgeo.2020.119853, 2020.
Fisher, B. J., Faust, J. C., Moore, O. M., Peacock, C. L., and März, C.: Electronic Annex for Technical Note: Uncovering the influence of methodological variations on the extractability of iron bound organic carbon Figshare [data set], https://doi.org/10.6084/m9.figshare.14384276, last access: 17 May 2021.
Hedges, J. I. and Keil, R. G.: Sedimentary organic matter preservation: an assessment and speculative synthesis, Mar. Chem., 49, 81–115, https://doi.org/10.1016/0304-4203(95)00008-f, 1995.
Hein, J. R., Koschinsky, A., Halbach, P., Manheim, F. T., Bau, M., Kang, J.-K., and Lubick, N.: Iron and manganese oxide mineralization in the Pacific, Geological Society, London, Special Publications, 119, 123, https://doi.org/10.1144/GSL.SP.1997.119.01.09, 1997.
Henkel, S., Kasten, S., Poulton, S. W., and Staubwasser, M.: Determination of the stable iron isotopic composition of sequentially leached iron phases in marine sediments, Chem. Geol., 421, 93–102, https://doi.org/10.1016/j.chemgeo.2015.12.003, 2016.
Hopkins, J.: JR16006 Cruise summary report, British Oceanographic Data Centre, available at: https://www.bodc.ac.uk/resources/inventories/cruise_inventory/reports/jr16006.pdf (last access: 17 May 2021), 2017.
Jelavić, S., Mitchell, A. C., and Sand, K. K.: Fate of organic compounds during transformation of ferrihydrite in iron formations, Geochem. Perspec. Lett., 15, 25–29, https://doi.org/10.7185/geochemlet.2030, 2020.
Karlsson, T. and Persson, P.: Coordination chemistry and hydrolysis of Fe(III) in a peat humic acid studied by X-ray absorption spectroscopy, Geochim. Cosmochim. Ac., 74, 30–40, https://doi.org/10.1016/j.gca.2009.09.023, 2010.
Karlsson, T. and Persson, P.: Complexes with aquatic organic matter suppress hydrolysis and precipitation of Fe(III), Chem. Geol., 322, 19–27, https://doi.org/10.1016/j.chemgeo.2012.06.003, 2012.
Keiser, L., Soreghan, G. S., and Joo, Y. J.: Effects Of Drying Techniques On Grain-Size Analyses of Fine-Grained Sediment, J. Sediment. Res., 84, 893–896, https://doi.org/10.2110/jsr.2014.68, 2014.
Kraal, P., Slomp, C. P., Reed, D. C., Reichart, G.-J., and Poulton, S. W.: Sedimentary phosphorus and iron cycling in and below the oxygen minimum zone of the northern Arabian Sea, Biogeosciences, 9, 2603–2624, https://doi.org/10.5194/bg-9-2603-2012, 2012.
Lalonde, K., Mucci, A., Ouellet, A., and Gelinas, Y.: Preservation of organic matter in sediments promoted by iron, Nature, 483, 198–200, https://doi.org/10.1038/nature10855, 2012.
Laufer, K., Michaud, A. B., Røy, H., and Jørgensen, B. B.: Reactivity of Iron Minerals in the Seabed Toward Microbial Reduction – A Comparison of Different Extraction Techniques, Geomicrobiol. J., 37, 170–189, https://doi.org/10.1080/01490451.2019.1679291, 2020.
Lem, W. and Wayman, M.: Decomposition of aqueous dithionite. Part I. Kinetics of decomposition of aqueous sodium dithionite, Can. J. Chem., 48, 776–781, 1970.
Lister, M. W. and Garvie, R. C.: Sodium Dithionite, Decomposition in Aqueous Solution and in the Solid State, Can. J. Chem., 37, 1567–1574, https://doi.org/10.1139/v59-228, 1959.
McClymont, E. L., Martínez-Garcia, A., and Rosell-Melé, A.: Benefits of freeze-drying sediments for the analysis of total chlorins and alkenone concentrations in marine sediments, Org. Geochem., 38, 1002–1007, https://doi.org/10.1016/j.orggeochem.2007.01.006, 2007.
McKeague, J. A. and Day, J. H.: Dithionite and oxalate extractable Fe and Al as aids in differentiating various classes of soil, Can. J. Soil Sci., 46, 13–22, https://doi.org/10.4141/cjss66-003, 1966.
Mehra, O. P. and Jackson, M. L.: Iron Oxide Removal from Soils and Clays by a Dithionite-Citrate System Buffered with Sodium Bicarbonate, Clay. Clay Miner., 7, 317–327, https://doi.org/10.1346/ccmn.1958.0070122, 1958.
Mikutta, C.: X-ray absorption spectroscopy study on the effect of hydroxybenzoic acids on the formation and structure of ferrihydrite, Geochim. Cosmochim. Ac., 75, 5122–5139, https://doi.org/10.1016/j.gca.2011.06.002, 2011.
Patzner, M. S., Mueller, C. W., Malusova, M., Baur, M., Nikeleit, V., Scholten, T., Hoeschen, C., Byrne, J. M., Borch, T., Kappler, A., and Bryce, C.: Iron mineral dissolution releases iron and associated organic carbon during permafrost thaw, Nat. Commun., 11, 6329, https://doi.org/10.1038/s41467-020-20102-6, 2020.
Poulton, S. W. and Canfield, D. E.: Co-diagenesis of iron and phosphorus in hydrothermal sediments from the southern East Pacific Rise: Implications for the evaluation of paleoseawater phosphate concentrations, Geochim. Cosmochim. Ac., 70, 5883–5898, https://doi.org/10.1016/j.gca.2006.01.030, 2006.
Raiswell, R., Canfield, D. E., and Berner, R. A.: A comparison of iron extraction methods for the determination of degree of pyritisation and the recognition of iron-limited pyrite formation, Chem. Geol., 111, 101–110, https://doi.org/10.1016/0009-2541(94)90084-1, 1994.
Raiswell, R. and Canfield, D. E.: Sources of iron for pyrite formation in marine sediments, Am. J. Sci., 298, 219–245, 1998.
Riedinger, N., Brunner, B., Krastel, S., Arnold, G. L., Wehrmann, L. M., Formolo, M. J., Beck, A., Bates, S. M., Henkel, S., Kasten, S., and Lyons, T. W.: Sulfur Cycling in an Iron Oxide-Dominated, Dynamic Marine Depositional System: The Argentine Continental Margin, Front. Earth Sci., 5, 33, https://doi.org/10.3389/feart.2017.00033, 2017.
Ruttenberg, K. C.: Development of a sequential extraction method for different forms of phosphorus in marine sediments, Limnol. Oceanogr., 37, 1460–1482, https://doi.org/10.4319/lo.1992.37.7.1460, 1992.
Salvadó, J. A., Tesi, T., Andersson, A., Ingri, J., Dudarev, O. V., Semiletov, I. P., and Gustafsson, Ö.: Organic carbon remobilized from thawing permafrost is resequestered by reactive iron on the Eurasian Arctic Shelf, Geophys. Res. Lett., 42, 8122–8130, https://doi.org/10.1002/2015gl066058, 2015.
Sirois, M., Couturier, M., Barber, A., Gélinas, Y., and Chaillou, G.: Interactions between iron and organic carbon in a sandy beach subterranean estuary, Mar. Chem., 202, 86–96, https://doi.org/10.1016/j.marchem.2018.02.004, 2018.
Slomp, C. P., Van der Gaast, S. J., and Van Raaphorst, W.: Phosphorus binding by poorly crystalline iron oxides in North Sea sediments, Mar. Chem., 52, 55–73, https://doi.org/10.1016/0304-4203(95)00078-X, 1996.
Thompson, J., Poulton, S. W., Guilbaud, R., Doyle, K. A., Reid, S., and Krom, M. D.: Development of a modified SEDEX phosphorus speciation method for ancient rocks and modern iron-rich sediments, Chem. Geol., 524, 383–393, https://doi.org/10.1016/j.chemgeo.2019.07.003, 2019.
van Bodegom, P. M., van Reeven, J., and Denier van der Gon, H. A. C.: Prediction of reducible soil iron content from iron extraction data, Biogeochemistry, 64, 231–245, https://doi.org/10.1023/a:1024935107543, 2003.
Wagai, R. and Mayer, L. M.: Sorptive stabilization of organic matter in soils by hydrous iron oxides, Geochim. Cosmochim. Ac., 71, 25–35, https://doi.org/10.1016/j.gca.2006.08.047, 2007.
Wehrmann, L. M., Formolo, M. J., Owens, J. D., Raiswell, R., Ferdelman, T. G., Riedinger, N., and Lyons, T. W.: Iron and manganese speciation and cycling in glacially influenced high-latitude fjord sediments (West Spitsbergen, Svalbard): Evidence for a benthic recycling-transport mechanism, Geochim. Cosmochim. Ac., 141, 628–655, https://doi.org/10.1016/j.gca.2014.06.007, 2014.
Zhang, D., Wang, S., Wang, Y., Gomez, M. A., Duan, Y., and Jia, Y.: The Transformation of Two-Line Ferrihydrite into Crystalline Products: Effect of pH and Media (Sulfate versus Nitrate), ACS Earth and Space Chemistry, 2, 577–587, https://doi.org/10.1021/acsearthspacechem.8b00001, 2018.
Zhang, S., Wang, S., and Shan, X.-Q.: Effect of sample pretreatment upon the metal speciation in sediments by a sequential extraction procedure, Chem. Spec. Bioavailab., 13, 69–74, https://doi.org/10.3184/095422901782775435, 2001.
Zhao, Q., Poulson, S. R., Obrist, D., Sumaila, S., Dynes, J. J., McBeth, J. M., and Yang, Y.: Iron-bound organic carbon in forest soils: quantification and characterization, Biogeosciences, 13, 4777–4788, https://doi.org/10.5194/bg-13-4777-2016, 2016.