the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 24 Mar 2025
| 24 Mar 2025
Dissolved organic matter fosters core mercury-methylating microbiomes for methylmercury production in paddy soils
Qiang Pu
Bo Meng
Jen-How Huang
Kun Zhang
Jiang Liu
Yurong Liu
Mahmoud A. Abdelhafiz
Xinbin Feng
Methylmercury (MeHg), accumulated in rice grains, is highly toxic for humans. Its production is largely driven by microbial methylation in paddy soils; however, dissolved organic matter (DOM) is a critical component of the soil biogeochemistry process, yet its interactions with microorganisms involved in MeHg production remain poorly understood. Here, we conducted hgcA gene sequencing and a genome-resolved metagenomic analysis to identify core Hg-methylating microbiomes and investigate the effect of DOM on core Hg-methylating microbiomes in paddy soils across a Hg contamination gradient. In general, the Hg-methylating microbial communities varied largely with the degree of Hg contamination in soils. Surprisingly, a core Hg-methylating microbiome was identified that was exclusively associated with MeHg concentration. The partial Mantel test revealed strong linkages among core Hg-methylating microbiome composition, DOM, and MeHg concentration. Structural equation models further indicated that core Hg-methylating microbiome composition significantly impacted soil MeHg concentration, contributing 89 % of the observed variation, while DOM plays a crucial role in determining core Hg-methylating microbiome composition, accounting for 65 %. These results suggested that DOM regulates MeHg production by altering the composition of core Hg-methylating microbiomes. The presence of various genes associated with carbon metabolism in the metagenome-assembled genome of core Hg-methylating microorganisms suggests that different DOM stimulates the activity of core Hg-methylating microorganisms to methylate Hg, which was confirmed by a pure incubation experiment with Geobacter sulfurreducens PCA (a core Hg-methylating microorganism) amended with a natural DOM solution extracted from investigated soils. Overall, DOM simultaneously changes core Hg-methylating microbiome composition and functional activity and thus enhances MeHg production in paddy soils.
- Article
(6279 KB) - Full-text XML
-
Supplement
(950 KB) - BibTeX
- EndNote




Mercury (Hg) is a toxic contaminant since it can be transformed into neurotoxic methylmercury (MeHg) and biomagnified in food chains (Driscoll et al., 2013). Human exposure to MeHg can cause neurocognitive deficits and cardiovascular effects (Oulhote et al., 2017; Roman et al., 2011). It is generally accepted that seafood consumption is the major route of exposure to MeHg in humans (Schartup et al., 2019). However, recent studies have demonstrated that rice consumption is another important route of human exposure to MeHg (Feng et al., 2008), with 3.5 billion individuals relying on rice as a principal dietary component (Muthayya et al., 2014).
Compared to other environments such as wetlands and aquatic sediments, paddy fields present unique ecological conditions that make them significant hotspots for Hg methylation. The frequent flooding and draining cycles, high organic matter content, and dynamic redox conditions in paddy soils create an environment that supports high levels of microbial activity, particularly Hg-methylating microorganisms (Yin et al., 2013). These conditions not only enhance MeHg production but also increase the likelihood of MeHg entering the food web through rice consumption, posing significant health risks (Zhang et al., 2010). Understanding Hg methylation in paddy fields is therefore crucial, as rice is a critical exposure route for MeHg in humans.
The accumulation of MeHg in rice is mostly attributed to microbial methylation of inorganic Hg in paddy soils (Meng et al., 2011). In situ methylation and demethylation are deemed to be important processes controlling the net MeHg concentration in environments (Barkay and Gu, 2022; Helmrich et al., 2021; Li and Cai, 2012). Our recent study showed that Hg transformation processes, such as methylation, demethylation, oxidation, and reduction, occurred simultaneously in paddy soils, with Hg methylation being the most active (Liu et al., 2023). Therefore, paddy soil is a typical “hotspot” for Hg methylation, which is mainly a biotic process mediated by many abiotic factors, such as Hg bioavailability and redox conditions (Li and Cai, 2012). The diversity and activity of Hg-methylating microorganisms in paddy soils control MeHg production (Gilmour et al., 2013; Liu et al., 2018). However, among the various Hg-methylating microorganisms currently known, the core microbiome controlling MeHg production and its interaction with environmental variables in paddy soils have yet to be identified.
Physicochemical factors in soils, such as organic matter, pH, salinity, redox potential, iron, and sulfur, have been shown to regulate the activity of Hg-methylating microorganisms and play an important role in controlling MeHg production in rice fields (Ullrich et al., 2001). Among the different variables, soil organic matter, which is ubiquitous in paddy soils (Li et al., 2018), plays a vital role in Hg methylation (Yin et al., 2013). Dissolved organic matter (DOM), the most mobile organic matter fraction, increases MeHg production under sulfidic conditions (Graham et al., 2012). DOM increases microbial Hg bioavailability for methylation by stabilizing β-HgS(s) nanoparticles to prevent aggregation. In addition, Hg speciation in Hg-polluted paddy soils was found to be predominantly regulated by organic matter (Liu et al., 2022), and the high bioavailability of DOM-bound Hg in rice paddies contributed to an increase in MeHg production (Liu et al., 2022). In contrast, other studies reported that DOM had a high affinity for Hg compounds (Skyllberg et al., 2006), suppressing MeHg production due to strong Hg–DOM complexation (Schartup et al., 2015). As a result, the role of paddy soil DOM on Hg methylation remains elusive. Our recent study showed a significant and strong relationship between MeHg production and low-molecular-weight DOM in paddy soils collected from major rice-producing areas across China (Abdelhafiz et al., 2023). Given paddy soil DOM's significant chemodiversity (Li et al., 2018), it is reasonable to hypothesize that the effect of DOM on MeHg production cannot be assessed solely based on Hg speciation and bioavailability, suggesting that other factors also play roles in MeHg production.
MeHg production is controlled by the synergy of Hg bioavailability and Hg-methylation capacity (Peterson et al., 2023), indicating that Hg-methylating microbial communities may also play an important role in DOM-regulated MeHg production. Concentration and composition of DOM have been shown to regulate MeHg production via alteration of the composition of the soil microbial community (Fagervold et al., 2014; Hu et al., 2021; Oloo et al., 2016). However, the core Hg-methylating microorganisms were not identified within these studies. Zhao et al. (2017) reported that two model Hg methylators exhibited an opposite response to DOM at the strain level. Therefore, we hypothesized that DOM fosters a core Hg-methylating microbiome that regulates MeHg production, since the core microbiome has a pivotal role in the functioning of ecosystems (Banerjee et al., 2018; Chen et al., 2019; Xun et al., 2021).
Thus, an attempt was made within this study to verify the crucial role of DOM in fostering the core Hg-methylating microbiome for MeHg production by (1) identifying the core Hg-methylating microbiome in paddy soils across a gradient of Hg contamination, (2) quantifying the relevance of DOM to the core Hg-methylating microbiome and MeHg production in paddy soils compared with other soil physicochemical parameters, and (3) elucidating the mechanism of core Hg-methylating microorganisms in response to different DOM. These results broaden our understanding of DOM as the prominent factor in altering Hg-methylating microbial communities and highlight the contribution of the core Hg-methylating microbiome to MeHg production in paddy soils.
2.1 Soil sampling and physicochemical analysis
Two field sampling campaigns were conducted in September 2020 and August 2022 in this study. Specifically, paddy fields from an abandoned Hg mining area (Sikeng, SK), an artisanal Hg smelting area (Gouxi, GX), and a regional background area (Huaxi, HX) in Guizhou Province, SW China, were selected in September 2020 (Tables S1, Samples S1–27). In each study area (SK, GX, and HX), nine sampling sites were randomly selected. Similarly, 19 additional sampling sites from the rice-producing areas in 12 provinces of China were selected in August 2022 (Table S1, Samples S28–46). At each site, one rice paddy field was randomly selected. Paddy soil was taken from the root zone (10–20 cm deep) and comprised a composite of three subsamples from the same paddy field. A total of 46 soil samples were obtained in this study to represent different Hg contamination levels and bioavailability, net MeHg production, DOM concentration and composition, soil microbial community composition and structure, and other physicochemical characteristics. Soil samples were collected in sterile PP bottles (Nalgene®, Thermo Fisher, USA) without any headspace, immediately shipped back to the laboratory on ice packs (∼ 4 °C), and divided into two subsamples before use. One subsample was stored at −20 °C for microbial analysis, and the other was stored at 4 °C for the analysis of soil physicochemical properties. Freeze-dried samples (−80 °C; EYELA FDU-2110, China) were screened to remove gravel and residue and then ground and evenly mixed using a mortar and pestle to pass through a 200-mesh sieve. The processed soil samples were analysed for pH, total carbon (TC), total nitrogen (TN), various mercury species (water-soluble Hg, total Hg (THg), and MeHg), water-soluble sulfate (SO) and nitrate (NO), DOM concentration (measured as water-soluble dissolved organic carbon), DOM composition (measured as optical properties of DOM), and low-molecular-weight organic acids. Fresh soil samples were also centrifuged to obtain pore water for the analysis of iron and sulfur (measured as Fe2+ and S2− in soil pore water). Detailed measurement procedures are provided in Supplement Text S1. It should be noted that Fe2+ and S2− data were limited to soil samples obtained in August 2022.
2.2 Soil DNA extraction
We extracted DNA from 0.5 g of soil using the FastDNA Spin Kit for Soil (MP Biomedicals, France), following the manufacturer's instructions. The quality and concentration of the isolated DNA were assessed using spectrophotometry (Nanodrop ND1000, USA) and 1.0 % agarose gel electrophoresis. The DNA was then stored at −80 °C for further analysis.
2.3 Amplicon sequencing and bioinformatic analysis
Soil Hg-methylating microbial communities were characterized by Illumina MiSeq sequencing of the hgcA gene using the primer pair ORNL-HgcAB-uni-F (5′-AAYGTCTGGTGYGCNGCVGG-3′) and the reverse primer ORNL-HgcAB-uni-32R (5′-CAGGCNCCGCAYTCSATRCA-3′) (Gionfriddo et al., 2020). Amplicons were equimolarly mixed and sequenced using the Illumina MiSeq instrument (Illumina Inc., San Diego) in 2×300 bp mode. Poor-quality reads, adapters, and primers were trimmed with Sickle and Cutadapt (Joshi and Fass, 2011; Martin, 2011). USEARCH (version 8.0) was used to truncate, dereplicate, sort, and remove singletons (Edgar, 2013). The set of sequences obtained was clustered at a 60 % similarity cutoff with cd-hit-est (Fu et al., 2012). Using USEARCH (version 8.0), the sequences were then mapped to the resulting clusters' representative sequences to build a count table. The sequences were annotated with amino acid sequences from Hg-MATE-Db (V1.01142021) (Gionfriddo et al., 2021) by using a hidden Markov model (HMM) based on HMMER (Eddy, 2011). In addition, the abundance of the Hg-methylating gene hgcA (which encodes a corrinoid protein essential for methylating inorganic Hg) was quantified in an Applied Biosystems 7500. The quantification of the hgcA gene is provided in Supplement Text S2.
2.4 Metagenomic sequencing and bioinformatic analysis
DNA from nine randomly selected paddy fields at each site in September 2020 was equimolarly mixed to obtain > 1 µg of DNA for shotgun metagenomic sequencing. For paddy soils collected in August 2022, three replicates of each sample were utilized to ensure sufficient quantity and quality of DNA for metagenomic sequencing. A total of 22 samples were analysed using an Illumina HiSeq 2500 system (Illumina Corp., USA).
The detection and taxonomic identification of the hgcAB gene (full operon responsible for Hg methylation pathway) were performed with marky-coco (Capo et al., 2023). The metagenomic sequences were trimmed to eliminate low-quality reads using fastp with the following parameters: -q30-l25 –detect_adapter_for_pe –trim_poly_g –trim_poly_x (Chen et al., 2018). These high-quality reads were then assembled into contigs using megahit 1.1.2 with default settings (Li et al., 2016). The annotation of the contigs for prokaryotic protein-coding gene prediction was conducted using prodigal 2.6.3 (Hyatt et al., 2010). To search for hgc homologs, a profile of HMM derived from Hg-MATE.db.v1 was applied to an amino acid FASTA file generated from each assembly with the function hmmsearch from HMMER 3.2.1 (Finn et al., 2011). To eliminate paralogs of hgcA, we removed the sequences without the conserved putative cap helix motif [N(V/I)WCA(A/G)GK] reported previously (Parks et al., 2013). We further filtered the sequences by retaining only sequences with more than four transmembrane domains as identified by TMHMM (v.2.0) (Krogh et al., 2001). Finally, the obtained contigs with hgcA homologs were classified taxonomically following a previously described method (Zhang et al., 2023). In addition, to estimate the relative abundance of the hgcA gene, metagenomic reads were mapped to representative genomes of the hgcA dataset using Bowtie2 (Capo et al., 2023). The relative abundances of each gene were calculated by normalizing the total length of successfully mapped reads by gene length and the total number of reads in the metagenome.
Contigs of ≥ 1000 bp were used to carry out a binning analysis with the MetaWRAP pipeline (v1.3.2) (Uritskiy et al., 2018). The quality of reconstructed metagenome-assembled genomes (MAGs) was assessed using CheckM (Parks et al., 2015). High-quality MAGs (completeness ≥ 90 % and contamination ≤ 10 %) were used to detect hgcA homologs, and taxonomy of these retrieved MAGs was conducted using GTDB-tk (v2.1.0) with its reference database (version release_207V2) (Parks et al., 2022). To explore what fractions of DOM can be metabolized by core Hg-methylating microorganisms, core Hg-methylating microbial-associated MAGs were mapped to the protein sequence of the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using the eggNOG-mapper (Huerta-Cepas et al., 2017).
2.5 Pure incubation of Geobacter sulfurreducens PCA with different DOM
To validate that different concentrations and molecular weights of DOM stimulate the activity of core Hg-methylating microorganisms, we incubated Geobacter sulfurreducens PCA (G. sulfurreducens PCA), identified as a core Hg-methylating microorganism in this study, with Hg2+, and a natural DOM solution extracted from non-Hg-polluted soil (NMS), moderately Hg-polluted soil (MMS), or highly Hg-polluted soil (HMS). Geobacter was selected for these pure incubation experiments due to its dominant role in mercury methylation and its ability to isolate the effects of DOM on methylation rates without the interference of soil matrix complexity. More details on the descriptions of the pure incubation experiment can be found in Supplement Text S3.
2.6 Statistical analysis
All statistical analyses were conducted using the R platform (version 3.6.1). All statistical tests were considered significant at p<0.05. The Kruskal–Wallis test was used to compare microbial alpha diversity among all samples. Hg-methylating microbial communities across differently polluted soils were compared by analysing dissimilarity matrices using the Bray–Curtis distance and visualized using principal coordinates analysis (PCoA) and Adonis with the ade4 and vegan packages (Dray and Dufour, 2007; Oksanen et al., 2017). To determine the relationship between THg and MeHg, Spearman correlation was performed using ggpubr and visualized using ggplot2 packages (Kassambara, 2018; Wickham, 2009). Variation partitioning analysis was performed using the vegan package (Oksanen et al., 2017). The major predictors of Hg-methylating microbial communities and their significance were identified using random forest analysis with randomForest, rfPermute, and A3 packages (Archer, 2018; Fortmann-Roe, 2015; Liaw and Wiener, 2002). To investigate the co-occurrence patterns among microbial taxa related to MeHg production, co-occurrence networks were established in the R platform using the psych package (Revelle, 2023), and visualized in Gephi 0.9.2 (Bastian et al., 2009) based on strong (Spearman's r>0.8) and significant (p<0.01) correlations (De Cáceres and Legendre, 2009). The modules in Hg-methylating microbial network were identified using default parameters from Gephi. To explore the relationship between the modules and environmental parameters, we correlated dissimilarities of bacterial composition in a core Hg-methylating microbiome with those of environmental factors as previously described (Sunagawa et al., 2015). The structural equation model (SEM) was conducted using the R platform to evaluate the impacts of DOM and a core Hg-methylating microbiome on MeHg production. A prior model was established based on the known relationships among drivers impacting MeHg production (Fig. S1). We further calculated the contribution of ecological parameters, including DOM, to the core Hg-methylating microbiome and the contribution of the core Hg-methylating microbiome to MeHg production, following the approach described by Tao et al. (2015). This calculation was performed by determining the proportion of the squared path coefficient of each parameter relative to the sum of the squared path coefficients of all parameters influencing the same target variable (Tao et al., 2015).
3.1 Mercury production in paddy soils
THg concentrations in paddy soils ranged from 0.03 to 1079.75 µg g−1 dry weight (dw) (Table S1). As reported in our previous study, dividing paddy soils by THg concentration rather than sampling sites facilitates a comprehensive investigation of the key factors influencing Hg methylation (Abdelhafiz et al., 2023). Therefore, the paddy soils in this study were divided into three categories according to THg concentration: non-Hg-polluted soil (NMS, with average levels of 0.24 ± 0.18 µg g−1 dw, n=23), moderately Hg-polluted soil (MMS, 18.28 ± 6.77 µg g−1 dw, n=13), and highly Hg-polluted soil (HMS, 637.79±160.93 µg g−1 dw, n=10). Furthermore, statistically significant differences in DOM concentrations (reflected by DOC concentration) and DOM composition (reflected by SR of DOM) were found in NMS, MMS, and HMS (Table S2). Specifically, DOC concentration varied significantly across the three soil types, with 0.48 ± 0.13 in NMS, 0.40 ± 0.07 in MMS, and 0.30 ± 0.10 in HMS. Similarly, the SR of DOM differed markedly between NMS (1.40 ± 0.76), MMS (0.89 ± 0.09), and HMS (0.46 ± 0.09). However, no discernible differences in physicochemical properties (e.g. pH, S2−, SO, NO, TN, TC, Fe2+) were observed in NMS, MMS, and HMS (Table S3).
In this study, we found MeHg concentrations in paddy soils in the order of HMS (5.01 ± 0.77 ng g−1 dw, n=10) > MMS (2.54 ± 0.72 ng g−1 dw, n=13) > NMS (0.76 ± 0.25 ng g−1 dw, n=23) (Fig. S2). Accordingly, a positive relationship was observed between total Hg and MeHg in different paddy soils (Fig. S3).
3.2 Core mercury-methylating microbiomes as predictors of MeHg production in paddy soils
Random forest results revealed that hgcA gene abundance, DOM concentration, DOM composition, water-soluble Hg, Fe2+, and S2− were significantly (p<0.05) associated with MeHg concentration (Fig. S4), with the hgcA gene the strongest predictor. The hgcA-gene-based taxonomic profiles of paddy soils reveal changes in Hg-methylating microbial community compositions across different levels of Hg pollution (Fig. 1a). Such observations were additionally supported by (1) the Chao1 index revealing the diversity of Hg-methylating microorganisms in the order of MMS (312.57 ± 44.73) > NMS (268.47 ± 81.85) > HMS (187.08 ± 131.62) (p<0.05; Fig. 1b) and (2) the divergent patterns of Hg-methylating microbial communities in paddy soils (p<0.01; Fig. 1c). The shotgun metagenomic results were consistent in detecting Hg-methylating microbial community composition and structure (Fig. S5). Proteobacteria, Acidobacteria, and Chloroflexi were the most abundant phyla in different paddy soils detected by both sequencing strategies. In summary, using both hgcA gene sequencing and metagenomic data, a significant difference in Hg-methylating microbial community structure and diversity was observed in paddy soils.
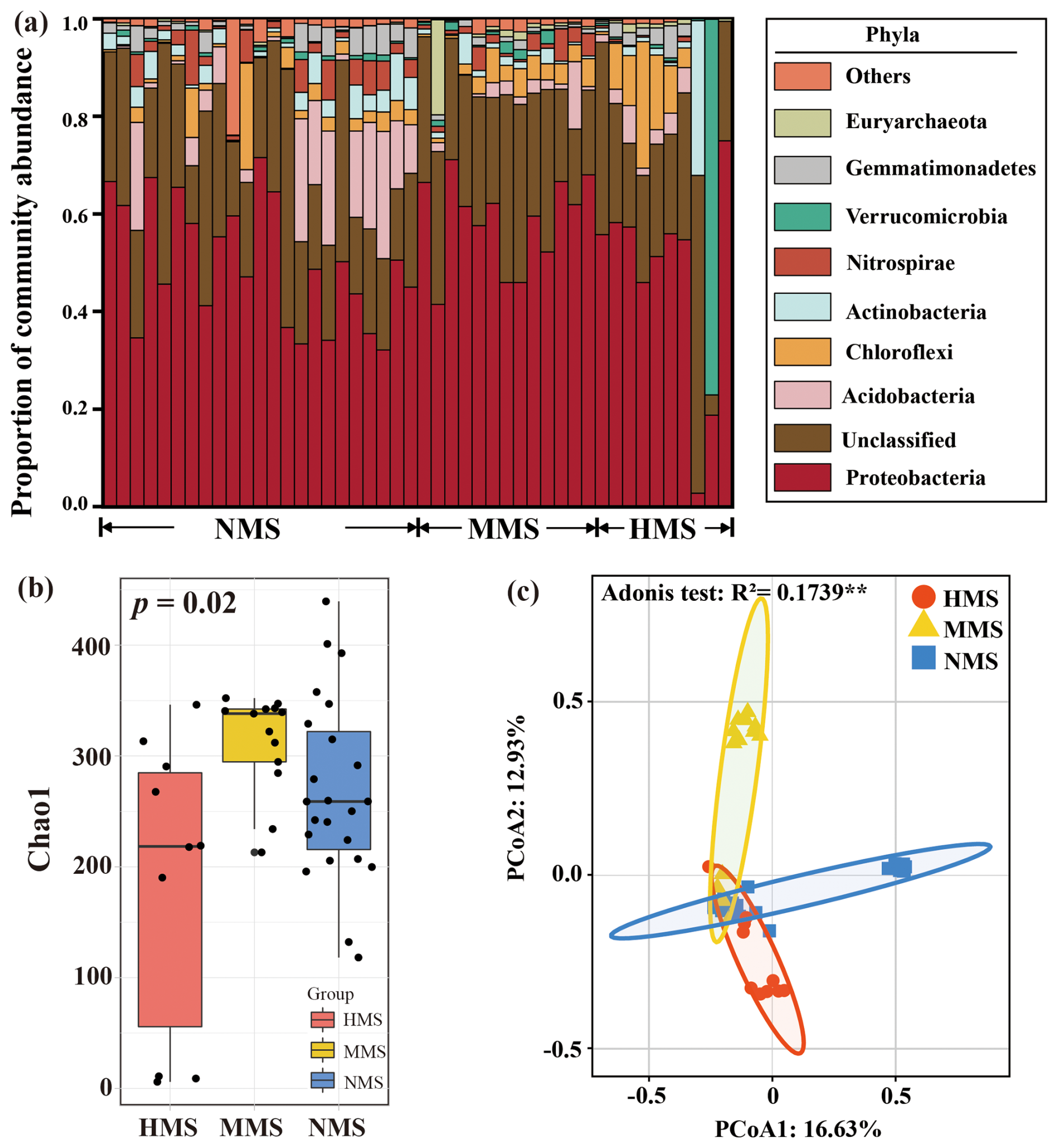
Figure 1Taxonomic profiles of Hg-methylating microbial communities in paddy soils based on amplicon sequencing. (a) Microbial community composition in differently polluted paddy soils. Phyla with low abundance grouped together under “other phyla”. (b) Microbial diversity (based on the Chao1 index) in differently polluted paddy soils. (c) Principal coordinates analysis (PCoA) based on the Bray–Curtis distance showing the overall pattern of Hg-methylating microbial communities in differently polluted paddy soils. NMS, non-Hg-polluted paddy soil (n=23); MMS, moderately Hg-polluted paddy soil (n=13); HMS, highly Hg-polluted paddy soil (n=10).
Network analysis captured 6, 11, and 11 modules (modularity index > 0.55) in NMS, MMS, and HMS, respectively (Fig. 2a, Table S4). Among all modules, Hg-methylating microorganisms in Module 1 in NMS, MMS, and HMS were identified as a core Hg-methylating microbiome based on their (1) higher connections to other modules and (2) higher abundance in total Hg-methylating microbial community (Table S5). Importantly, the impact of various modules in the microbial community on MeHg production was analysed using random forest analysis. The results revealed that the microbiome in Module 1 is a crucial bacterial group influencing soil methylmercury concentration (Fig. 2b). This group is considered the core Hg-methylating microbiome in this study. Further analysis of the core Hg-methylating microbiome composition revealed diverse core Hg-methylating microorganisms in paddy soils. Although most microorganisms are not annotated, the three genera with the highest abundance in each soil type are as follows: in NMS, Geobacter (36.2 %), Syntrophus (1.7 %), and Desulfomonas (0.4 %) dominate; in MMS, Geobacter (40.5 %), Granulicella (2.9 %), and Olavius (2.9 %) are the most abundant; and in HMS, Geobacter (56.8 %), Methanoregula (0.6 %), and Granulicella (2.3 %) prevail (Fig. 2c). It is worth highlighting that, in this study, microorganisms belonging to Geobacter were identified as the most significant core microorganisms for Hg methylation across all paddy soils.
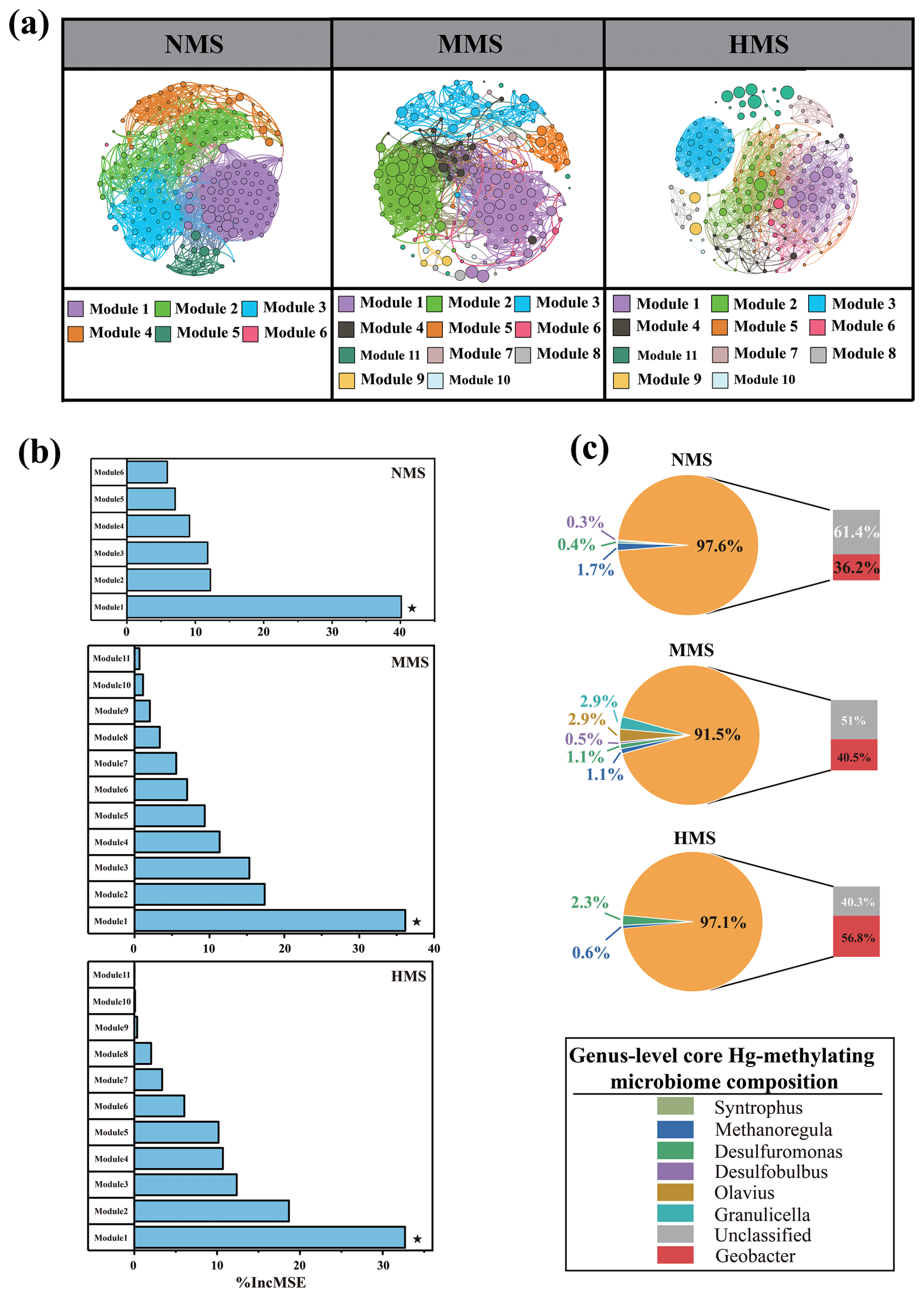
Figure 2Core Hg-methylating microbiomes in paddy soils. (a) Co-occurrence network of the Hg-methylating microbial community in differently polluted paddy soils. Each node represents 1 OTU. The node size is proportional to the relative abundance of OTUs. (b) Predictors of the MeHg production in differently polluted paddy soils based on random forest analysis. Only predictors with significant effects are denoted by asterisks. (c) Core Hg-methylating microbiome composition at genus level in differently polluted paddy soils. NMS, non-Hg-polluted paddy soil (n=23); MMS, moderately Hg-polluted paddy soil (n=13); HMS, highly Hg-polluted paddy soil (n=10).
3.3 Dissolved organic matter as indicators of core mercury-methylating microbiome composition in paddy soils
Based on an analysis of correlations, the results showed that there were significant correlations between core Hg-methylating microbiome composition, MeHg concentration, DOM concentration, DOM composition, water-soluble Hg, soil S2−, and Fe2+ (Fig. 3). Among all parameters, DOM is the most important factor influencing the composition of the core Hg-methylating microbiome. This was supported by DOM explaining most core Hg-methylating microbiome composition (Fig. S6). Random forest analysis also showed that DOM concentration and composition were the most important predictors of the composition of the core Hg-methylating microbiome (Fig. S7). Additionally, SEM results showed that the core Hg-methylating microbiome composition, which is closely linked to hgcA gene abundance, significantly regulated the soil MeHg concentration (λ=0.84, p<0.001) (Fig. 4). In comparison, the contributions of Hg bioavailability and redox conditions to the core Hg-methylating microbiome composition are 10 % and 25 %, respectively, which are much lower than that of DOM (65 %) (Fig. 4).
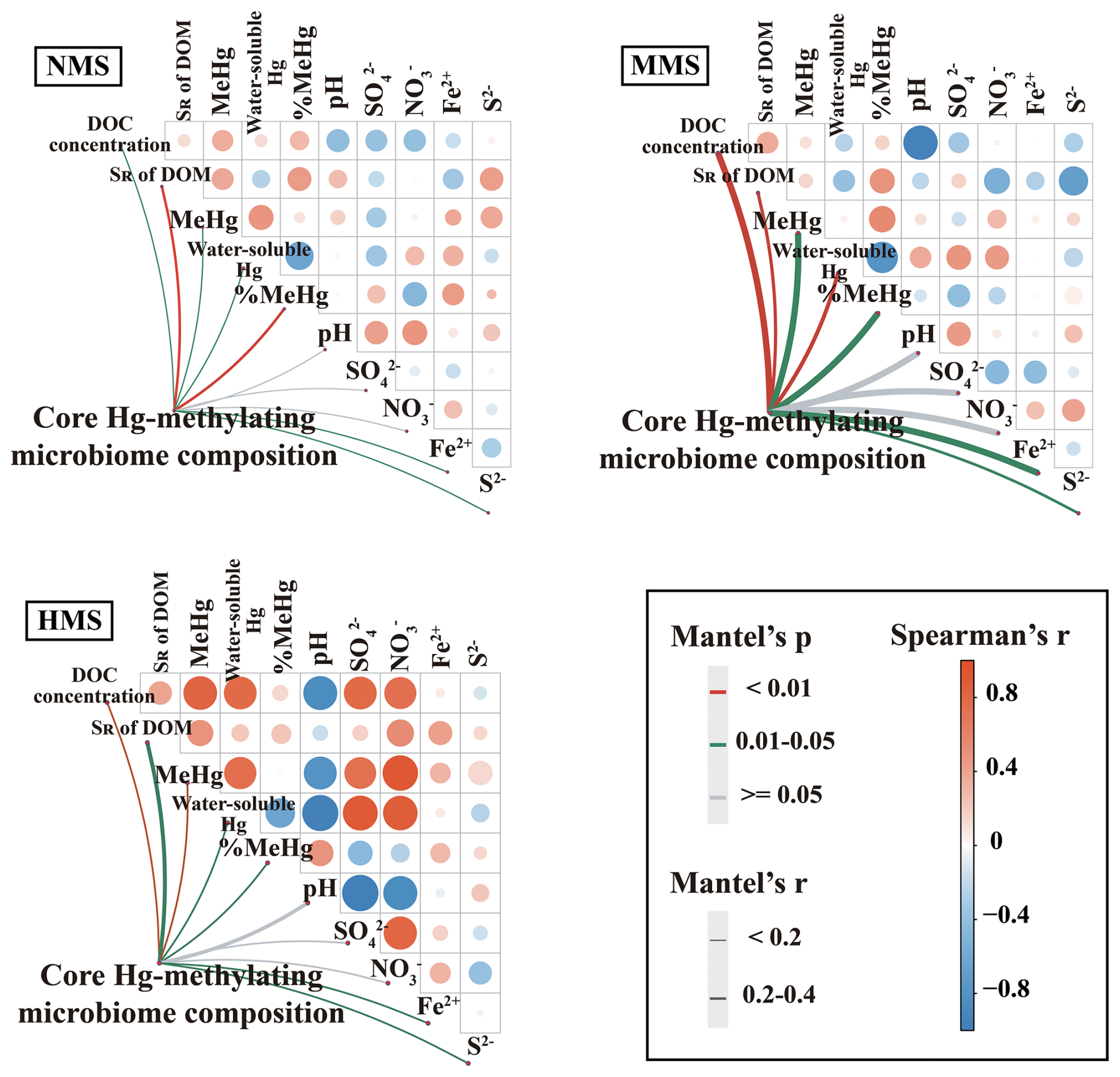
Figure 3Pairwise comparisons of environmental factors and community taxonomic composition in the core Hg-methylating microbiome in differently polluted paddy soils. NMS, non-Hg-polluted paddy soil; MMS, moderately Hg-polluted paddy soil; HMS, highly Hg-polluted paddy soil.
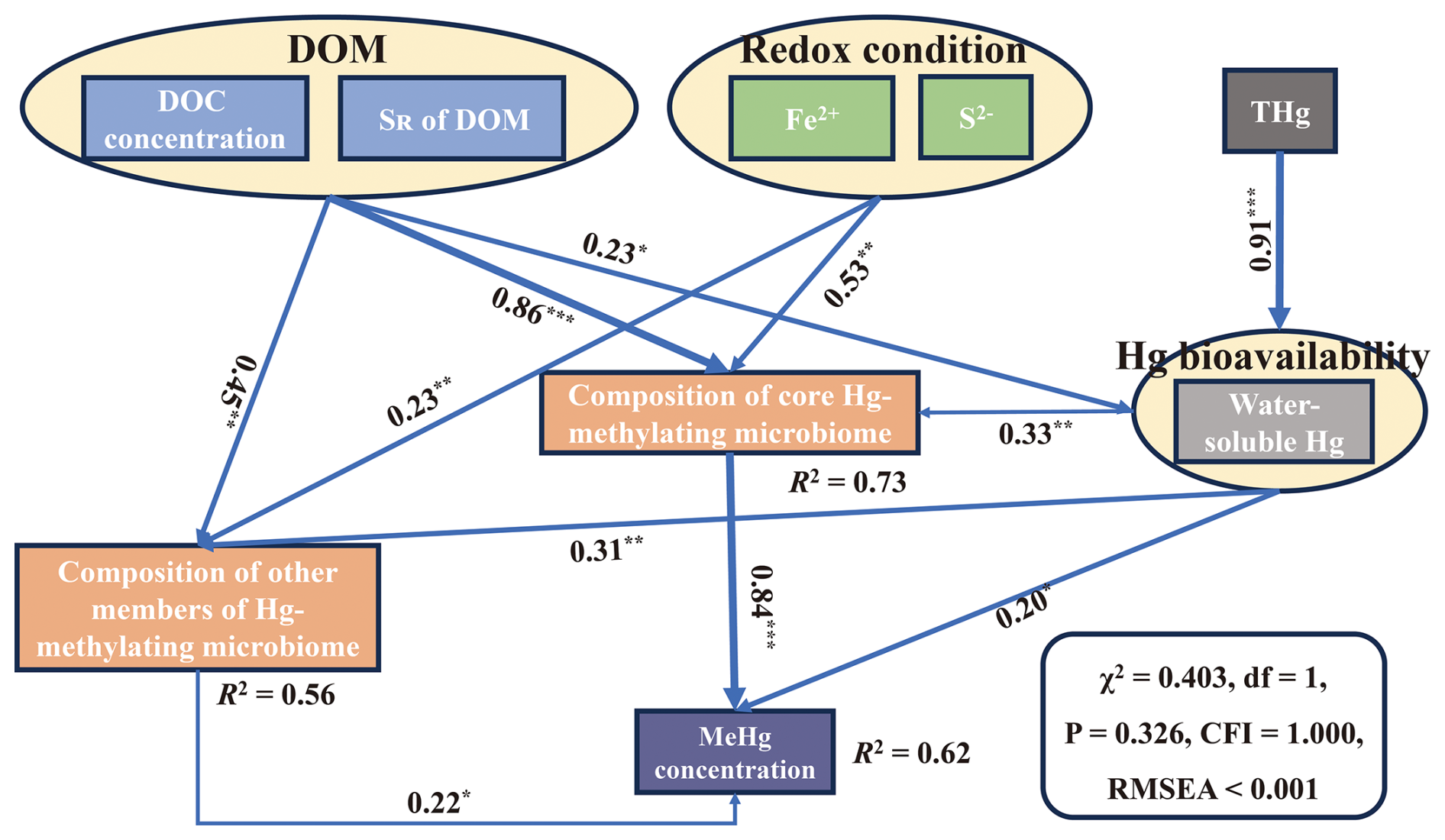
Figure 4Structural equation models showing the effects of DOM, redox conditions, and Hg bioavailability on MeHg production. NMDS1 values of the non-metric multidimensional scaling (NMDS) analysis were used for the representation of DOM and redox conditions in the SEMs. Numbers adjacent to arrows are standardized path coefficients, and numbers in brackets denote p values. “Statistically nonsignificant” results are not shown in the figure. R2 denotes the proportion of variance explained.
3.4 Dissolved organic matter stimulates activity of core mercury-methylating microorganisms, enhancing methylmercury production in paddy soils
The results of metagenomic binning revealed that three core Hg-methylating microbial-associated metagenome-assembled genomes (MAGs; completeness ≥ 90 % and contamination ≤ 10 %) carried different carbon utilization genes (ackA, sdhA, or ppdK genes) (Fig. 5), which are responsible for acetate kinase, succinate dehydrogenase, and pyruvate and orthophosphate dikinase. These results indicated that the low-molecular-weight DOM in soil selectively stimulates the activity of core Hg-methylating microorganisms that preferentially utilize them for metabolism, leading to the increase of MeHg concentration.
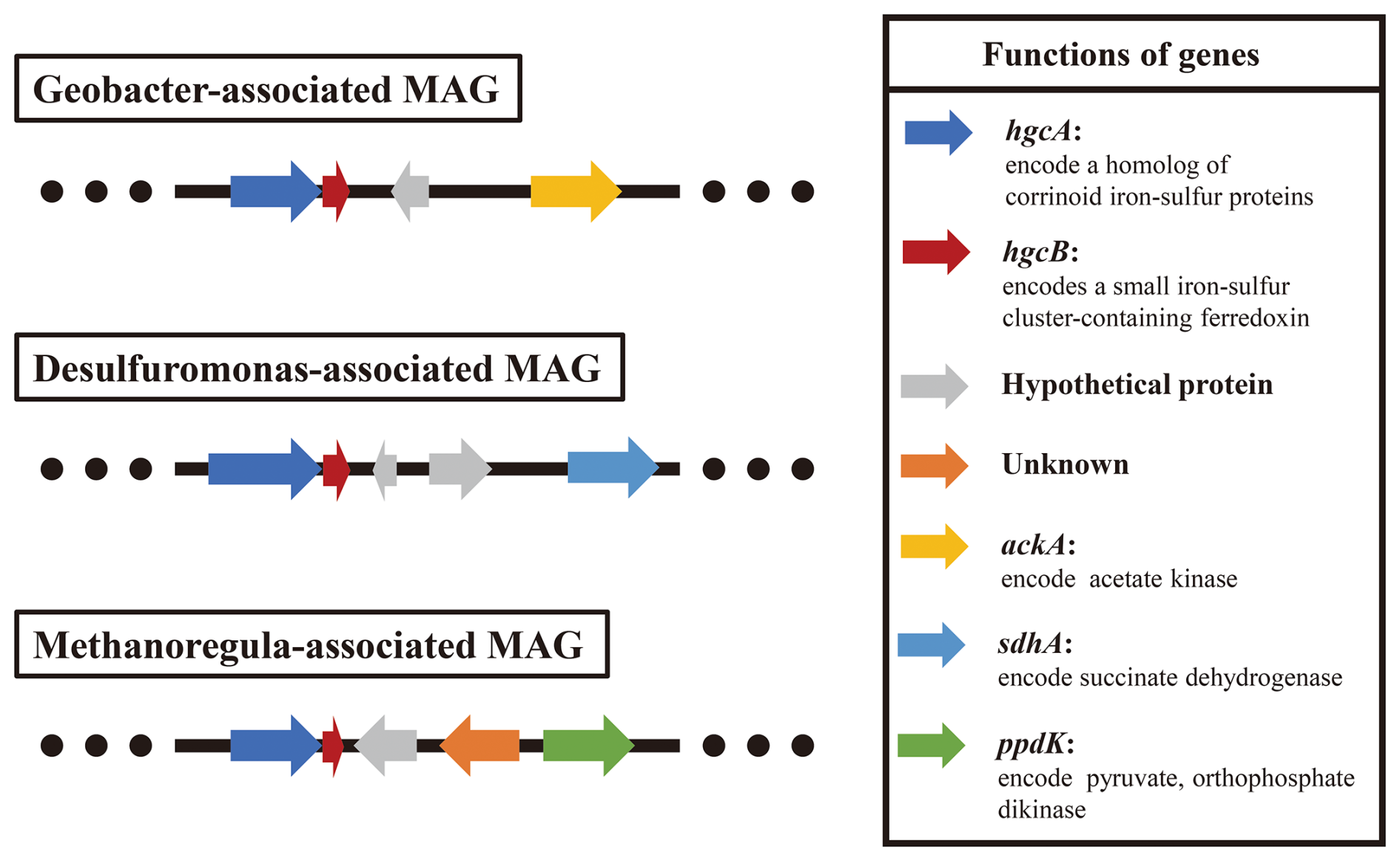
Figure 5Analysis of the genetic context of the hgcA gene and genes involved in carbon metabolism in core Hg-methylating microbial-associated MAGs. The extents and directions of genes are shown by arrows labelled with gene names.
To validate this hypothesis, Geobacter sulfurreducens PCA, a core Hg-methylating microorganism identified in this study, was incubated with HgCl2 and various DOM solutions extracted from investigated paddy soils. The results showed distinct patterns in MeHg production (Fig. 6), confirming that different concentrations of low-molecular-weight DOM significantly regulate MeHg production by influencing the activity of core Hg-methylating microorganisms.
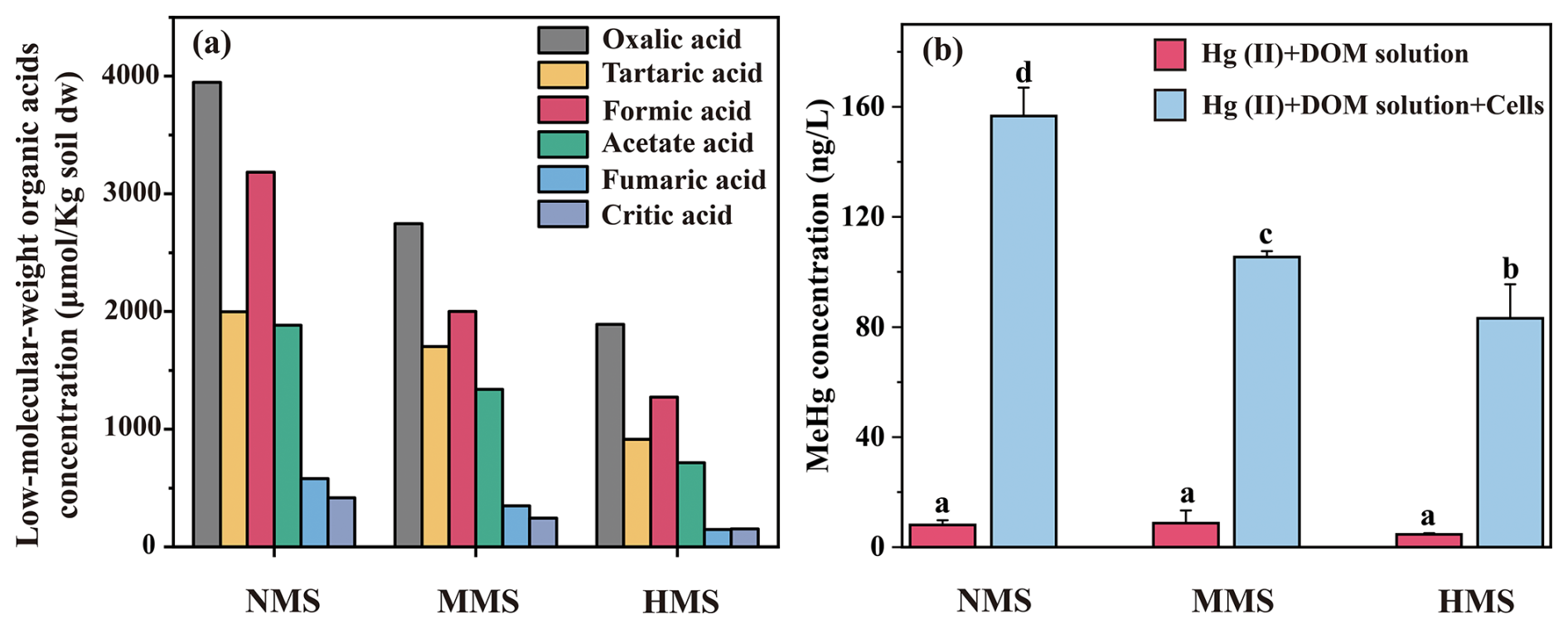
Figure 6Effect of natural DOM solution extracted from paddy soils on MeHg production by a core Hg methylator (Geobacter sulfurreducens PCA). (a) The concentration of low-molecular-weight organic acids in paddy soils from non-Hg-polluted soil (NMS), moderately Hg-polluted soil (MMS), and highly Hg-polluted soil (HMS). (b) MeHg concentration by G. sulfurreducens PCA. Data (n=3) are presented as the mean value ± SD, with error bars representing standard deviations. Significant differences among different treatments were tested with Tukey's honest significance test; different lowercase letters in each bar indicate significant differences among treatments (p<0.05).
Our study found that MeHg concentration was strongly linked to hgcA gene abundance even compared to abiotic factors, which suggested that MeHg production is a microbially mediated process (Parks et al., 2013; Podar et al., 2015). Our study further revealed that although there are significant differences in the Hg-methylating microbial communities in differently polluted paddy soils, they all have a core Hg-methylating microbiome, which plays a more important role than other Hg methylators in regulating MeHg production. As illustrated by a previous study, the major module (also known as the core microbiome) in a microbial community network contributes to the stability of a soil microbiome, enhancing its resistance to climate changes and nutrient fertilization (Jiao et al., 2022). These findings establish the presence of a major module contributing exclusively to Hg methylation in paddy soils, although there are many more Hg-methylating microorganisms present. In fact, microorganisms containing the hgcA gene are able to methylate Hg, but this does not mean that they are automatically active in Hg methylation.
The SEM analysis result indicated that although redox conditions and Hg bioavailability significantly affected the composition of a core Hg-methylating microbiome, their contribution to the composition of a core Hg-methylating microbiome was less and weaker than that of DOM. The explanation for this phenomenon may be the following:
-
The soil collected in the paddy field during the flooding period is in an anaerobic state, so the selection of redox conditions on core mercury-methylating microorganisms is weakened.
-
Hg is a toxic element to microorganisms and is usually not involved in microbial metabolism (Wang et al., 2020). Environmental Hg may induce the persistence of some microorganisms. Therefore, long-term Hg contamination often only elevates the abundance of specific microbial taxa capable of Hg tolerance (Frossard et al., 2018).
-
DOM, an important carbon source and nutrient in nature, is involved in microbial respiration and metabolism (Kujawinski, 2011). Consequently, the concentration and composition of DOM contributed significantly to core Hg-methylating microbiomes.
These results highlight the dominant role of DOM in shaping core Hg-methylating communities, as compared to redox conditions and Hg bioavailability.
Our study found that Geobacter, Desulfuromonas, Methanoregular, Syntrophus, Granulicella, and Olavius are core Hg-methylating microorganisms in paddy soils. Previous studies confirmed that Geobacter, Desulfuromonas, and Syntrophus have the capability for Hg methylation (Bravo et al., 2018; Gilmour et al., 2013; Liu et al., 2018; Zhong et al., 2024). In addition, Methanoregular spp., as methanogenic archaea, show potential for Hg methylation (Jones et al., 2019). Granulicella affects the decomposition of complex organic materials (Pankratov and Dedysh, 2010), while Olavius plays a role in sulfur and nitrogen cycling (Blazejak et al., 2005). These roles suggest that both microorganisms could also be important potential Hg methylators. Although many core Hg-methylating microorganisms have not been annotated, our study emphasizes that the annotated Hg-methylating microorganisms play a much greater role in Hg methylation in paddy soils than previously thought.
Our study identified various DOM components, including oxalic acid, tartaric acid, formic acid, acetate acid, fumaric acid, and citric acid, in paddy soils. These low-molecular-weight organic acids, particularly abundant in NMS soils, serve as key carbon sources for Hg-methylating microorganisms and stimulate the growth and activity of the core Hg-methylating microbiome. Pure incubation of Geobacter sulfurreducens PCA (a core Hg-methylating microorganism identified in our paddy soils) further confirmed that different concentrations of low-molecular-weight DOM solutions extracted from natural paddy soils obtained from NMS, MMS, and HMS had significant effects on MeHg concentration. These findings demonstrate that DOM composition strongly influences microbial Hg methylation by stimulating key metabolic pathways. For instance, Geobacter sulfurreducens and Desulfovibrio desulfuricans use acetate and fumarate in the TCA cycle, supporting anaerobic respiration and electron transport that enhance Hg methylation (Hu et al., 2013; Liu et al., 2018). Similarly, methanogenic archaea such as Methanoregula and Methanosarcina utilize formate and acetate through methanogenesis, further contributing to Hg methylation (Sakai et al., 2010; Schöne et al., 2022). Although metabolomic data were not included in this study, future research incorporating such analyses could provide valuable insights into how specific DOM components influence microbial metabolism and Hg methylation, revealing key metabolites and pathways such as acetate fermentation, methanogenesis, and electron transfer processes. This highlights how specific DOM components shape the core Hg-methylating microbiome and influence its role in MeHg production.
In contrast to low-molecular-weight organic acids, other DOM components such as aromatic compounds and humic substances may have limited influence on microbial Hg methylation due to their complex structures and reduced bioavailability. While aromatic compounds and humic substances were not directly analysed in this study, their complex structures likely reduce Hg bioavailability or slow microbial degradation, resulting in weaker effects on Hg methylation compared to low-molecular-weight organic acids. Future research could integrate direct Hg speciation measurements with detailed DOM compositional analyses to better understand how specific DOM components and Hg species interact to influence microbial Hg methylation.
DOM's influence on microbial Hg methylation has been observed in other ecosystems, such as wetlands and sediments, where DOM shapes microbial community structures to promote methylmercury (MeHg) production. For instance, in wetlands, DOM-bound Hg has been found to change the community assembly of mercury for methylating microbes (Fagervold et al., 2014). This highlights the broader ecological significance of DOM's role in promoting Hg methylation and suggests that DOM-driven microbiome modulation is a critical process across diverse environments. Moreover, the knowledge gained in this study highlights how variation in DOM quality due to human activities and climate change (e.g. changes in molecular weight, aromaticity, and bioactivity) could significantly alter MeHg production in different environmental compartments (Xenopoulos et al., 2021). For instance, long-term processes may scatter stable DOM, such as black carbon, globally through biomass combustion (Qi et al., 2020), while simpler and more reactive DOM may dominate in aquatic ecosystems (Xenopoulos et al., 2021). These changes could either enhance or diminish Hg ecotoxicity, depending on the specific conditions. Therefore, future in-depth studies coupling DOM quality, Hg speciation, and microbial Hg methylation are essential to deliver more accurate assessments of Hg's environmental and health impacts, particularly in the context of the Minamata Convention.
This study provides novel evidence that DOM significantly influences MeHg production by altering the composition and stimulating the activity of the core Hg-methylating microbiome. While DOM regulates the composition of other members of the Hg-methylating microbiome, its impact on MeHg production is primarily mediated through the core Hg-methylating microbiome. Using metagenomic binning and pure incubation experiments, we demonstrated that low-molecular-weight DOM directly promotes MeHg production by enhancing the metabolic activity of core Hg-methylating microorganisms. These findings underscore the central role of the core Hg-methylating microbiome in Hg cycling and highlight DOM as a critical driver of microbial Hg methylation. As human activities and climate change continue to alter DOM composition and concentration, their influence on Hg methylation dynamics warrants further investigation to better predict and mitigate Hg-related environmental and health risks.
The raw reads of hgcA gene amplicon sequencing have been deposited in the NCBI SRA under accession numbers PRJNA847325 and PRJNA972506. Shotgun metagenomic sequences have been deposited in the NCBI SRA under accession numbers PRJNA848068 and PRJNA972502. Other datasets generated during the current study are available from the corresponding author upon reasonable request.
The supplement related to this article is available online at https://doi.org/10.5194/bg-22-1543-2025-supplement.
The study was designed by QP, BM, and XBF. QP, JL, and YRL conducted the sampling, performed the DNA extraction, and carried out the bioinformatic analyses. JHH, KZ, and MA performed the geochemical analyses. The manuscript was written by QP and BM, with assistance and input from co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We appreciate Alexandre J. Poulain (University of Ottawa, Canada) for his valuable advice on manuscript writing. We also appreciate Tao Jiang (Southwest University, China) for his important help in the analysis of natural organic matter. Our deep appreciation goes to Peng Liang (Zhejiang Agriculture and Forestry University) for generously providing the G. sulfurreducens PCA. Additionally, we are grateful to Ji Chen, Kun Kong, Qianshuo Zhang, Muhammad Wajahat Aslam, for their help with sample collection and measurements.
This research has been supported by the National Natural Science Foundation of China (grant nos. 41931297 and 42207164) and the Guizhou Provincial Science and Technology Department (grant no. Qian-Ke-He-Ji-Chu ZK [2022] Yi-Ban 566).
This paper was edited by Jianming Xu and reviewed by Emi Stuart and two anonymous referees.
Abdelhafiz, M. A., Liu, J., Jiang, T., Pu, Q., Aslam, M. W., Zhang, K., Meng, B., and Feng, X.: DOM influences Hg methylation in paddy soils across a Hg contamination gradient, Environ. Pollut., 322, 121237, https://doi.org/10.1016/j.envpol.2023.121237, 2023.
Archer, E.: rfPermute: estimate permutation p-values for random forest importance metrics, CRAN, https://CRAN.R-project.org/package=rfPermute (last access: 5 June 2024), 2018.
Banerjee, S. K., Schlaeppi, K., and van der Heijden, M. G. A.: Keystone taxa as drivers of microbiome structure and functioning, Nat. Rev. Microbiol., 16, 567–576, https://doi.org/10.1038/s41579-018-0024-1, 2018.
Barkay, T. and Gu, B.: Demethylation-The Other Side of the Mercury Methylation Coin: A Critical Review, ACS Environ. Au., 2, 77–97, https://doi.org/10.1021/acsenvironau.1c00022, 2022.
Bastian, M., Heymann, S., and Jacomy, M.: Gephi: An Open Source Software for Exploring and Manipulating Networks, In Proceedings of the Third International ICWSM conference (San Jose, California, USA, May 17–20, 2009), 361–362, AAAI Press, Article ID 154, ISBN 978-1-57735-431-3, https://doi.org/10.3610/icwsm.v3i1.13984, 2009.
Blazejak, A., Erséus, C., Amann, R., and Dubilier, N.: Coexistence of bacterial sulfide oxidizers, sulfate reducers, and spirochetes in a gutless worm (Oligochaeta) from the Peru margin, Appl. Environ. Microbiol., 71, 1553–1561, 2005.
Bravo, A. G., Zopfi, J., Buck, M., Xu, J., Bertilsson, S., Schaefer, J. K., Poté, J. W., and Cosio, C.: Geobacteraceae are important members of mercury-methylating microbial communities of sediments impacted by waste water releases, ISME J., 12, 802–812, https://doi.org/10.1038/s41396-017-0007-7, 2018.
Capo, E., Peterson, B. D., Kim, M., Jones, D. S., Acinas, S. G., Amyot, M., Bertilsson, S., Björn, E., Buck, M., Cosio, C., Elias, D. A., Gilmour, C. C., Goñi Urriza, M. S., Gu, B., Lin, H., Liu, Y., McMahon, K. D., Moreau, J. W., Pinhassi, J., Podar, M., Puente-Sánchez, F., Sánchez, P., Storck, V., Tada, Y., Vigneron, A., Walsh, D. A., Vandewalle-Capo, M., Bravo, A. G., and Gionfriddo, C. M.: A consensus protocol for the recovery of mercury methylation genes from metagenomes, Mol. Ecol. Resour., 23, 190–204, https://doi.org/10.1111/1755-0998.13687, 2023.
Chen, L., Jiang, Y., Liang, C., Luo, Y., Xu, Q., Han, C., Zhao, Q. G., and Sun, B.: Competitive interaction with keystone taxa induced negative priming under biochar amendments, Microbiome, 7, 77, https://doi.org/10.1186/s40168-019-0693-7, 2019.
Chen, S., Zhou, Y., Chen, Y., and Gu, J.: fastp: an ultra-fast all-in-one FASTQ preprocessor, Bioinformatics, 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560, 2018.
De Cáceres, M. and Legendre, P.: Associations between species and groups of sites: indices and statistical inference, Ecology, 90, 3566–3574, https://doi.org/10.1890/08-1823.1, 2009.
Dray, S. and Dufour, A. B.: The ade4 Package: Implementing the Duality Diagram for Ecologists, J. Stat. Softw., 22, 1–20, https://doi.org/10.18637/jss.v022.i04, 2007.
Driscoll, C. T., Mason, R. P., Chan, H. M., Jacob, D. J., and Pirrone, N.: Mercury as a Global Pollutant: Sources, Pathways, and Effects, Environ. Sci. Technol., 47, 4967–4983, https://doi.org/10.1021/es305071v, 2013.
Eddy, S. R.: Accelerated Profile HMM Searches, Plos. Comput. Biol., 7, e1002195, https://doi.org/10.1371/journal.pcbi.1002195, 2011.
Edgar, R. C.: UPARSE: highly accurate OTU sequences from microbial amplicon reads, Nat. Methods, 10, 996–998, https://doi.org/10.1038/nmeth.2604, 2013.
Fagervold, S. K., Bourgeois, S., Pruski, A. M., Charles, F., Kerhervé, P., Vétion, G., and Galand, P. E.: River organic matter shapes microbial communities in the sediment of the Rhône prodelta, ISME J., 8, 2327–2338, https://doi.org/10.1038/ismej.2014.86, 2014.
Feng, X., Li, P., Qiu, G., Wang, S. L., Li, G. H., Shang, L. H., Meng, B., jiang, H. W., Bai, W. Y., Li, Z. G., and Fu, X. W.: Human Exposure To Methylmercury through Rice Intake in Mercury Mining Areas, Guizhou Province, China, Environ. Sci. Technol., 42, 326–332, https://doi.org/10.1021/es071948x, 2008.
Finn, R. D., Clements, J., and Eddy, S. R.: HMMER web server: interactive sequence similarity searching, Nucleic Acids Res., 39, W29–W37, https://doi.org/10.1093/nar/gkr367, 2011.
Fortmann-Roe, S.: Consistent and Clear Reporting of Results from Diverse Modeling Techniques: The A3 Method, J. Stat. Softw., 66, 1–23, https://doi.org/10.18637/jss.v066.i07, 2015.
Frossard, A., Donhauser, J., Mestrot, A., Gygax, S., Bååth, E., and Frey, B.: Long- and short-term effects of mercury pollution on the soil microbiome, Soil. Biol. Biochem., 120, 191–199, https://doi.org/10.1016/j.soilbio.2018.01.028, 2018.
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W.: CD-HIT: accelerated for clustering the next-generation sequencing data, Bioinformatics, 28, 3150–3152, https://doi.org/10.1093/bioinformatics/bts565, 2012.
Gilmour, C. C., Podar, M., Bullock, A. L., Graham, A. M., Brown, S. D., Somenahally, A. C., Johs, A., Hurt, R. A., Bailey, K. L., and Elias, D. A.: Mercury methylation by novel microorganisms from new environments, Environ. Sci. Technol., 47, 11810–11820, https://doi.org/10.1021/es403075t, 2013.
Gionfriddo, C., Capo, E., Peterson, B., Heyu, L., Jones, D., Bravo, A. G., Bertilsson, S., Moreau, J., McMahon, K., Elias, D., and Gilmour, C.: Hg-MATEDb, v1.01142021, figshare, https://smithsonian.figshare.com/articles/dataset/Hg-MATE_Db_v1_01142021/13105370 (last access: 16 April 2024), https://doi.org/10.6084/m9.figshare.13105370, 2021.
Gionfriddo, C. M., Wymore, A. M., Jones, D. S., Wilpiszeski, R. L., Lynes, M. M., Christensen, G. A., Soren, A., Gilmour, C. C., Podar, M., and Elias, D. A.: An Improved hgcAB Primer Set and Direct High-Throughput Sequencing Expand Hg-Methylator Diversity in Nature, Front Microbiol., 11, 541554, https://doi.org/10.3389/fmicb.2020.541554, 2020.
Graham, A. M., Aiken, G. R., and Gilmour, C. C.: Dissolved organic matter enhances microbial mercury methylation under sulfidic conditions, Environ. Sci. Technol., 46 5, 2715–2723, https://doi.org/10.1021/es203658f, 2012.
Helmrich, S., Vlassopoulos, D., Alpers, C. N., and O'Day, P. A.: Critical review of mercury methylation and methylmercury demethylation rate constants in aquatic sediments for biogeochemical modeling, Crit. Rev. Env. Sci. Tec., 52, 4353–4378, https://doi.org/10.1080/10643389.2021.2013073, 2021.
Hu, H., Umbreen, S., Zhang, Y., Bao, M., Huang, C., and Zhou, C.: Significant association between soil dissolved organic matter and soil microbial communities following vegetation restoration in the Loess Plateau, Ecol. Eng., 169, 106305, https://doi.org/10.1016/j.ecoleng.2021.106305, 2021.
Hu, H., Lin, H., Zheng, W., Tomanicek, S. J., Johs, A., Feng, X., Elias, D. A., Liang, L., and Gu, B.: Oxidation and methylation of dissolved elemental mercury by anaerobic bacteria, Nat. Geosci., 6, 751–754, https://doi.org/10.1038/ngeo1894, 2013.
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., and Bork, P.: Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper, Mol. Biol. Evol., 34, 2115–2122, https://doi.org/10.1093/molbev/msx148, 2017.
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J.: Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification, BMC Bioinform., 11, 119, https://doi.org/10.1186/1471-2105-11-119, 2010.
Jiao, S., Qi, J., Jin, C., Liu, Y., Wang, Y., Pan, H., Chen, S., Liang, C., Peng, Z., Chen, B., Qian, X., and Wei, G.: Core phylotypes enhance the resistance of soil microbiome to environmental changes to maintain multifunctionality in agricultural ecosystems, Glob. Change Biol., 28, 6653–6664, https://doi.org/10.1111/gcb.16387, 2022.
Jones, D. S., Walker, G. M., Johnson, N. W., Mitchell, C. P. J., Coleman Wasik, J. K., and Bailey, J. V.: Molecular evidence for novel mercury methylating microorganisms in sulfate-impacted lakes, ISME J., 13, 1659–1675, https://doi.org/10.1038/s41396-019-0376-1, 2019.
Joshi, N. A. and Fass, J. N. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files, GitHub, https://github.com/najoshi/sickle (last access: 11 April 2024), 2011.
Kassambara, A.: ggpubr.: “ggplot2” based publication ready plots, R package version 0.2, https://CRAN.R-project.org/package=ggpubr (last access: 24 April 2024), 2018.
Krogh, A., Larsson, B., Heijne, G. V., and Sonnhammer, E. L. L.: Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes, J. Mol. Biol., 305, 567–580, https://doi.org/10.1006/jmbi.2000.4315, 2001.
Kujawinski, E. B.: The impact of microbial metabolism on marine dissolved organic matter, Annu. Rev. Mar., 3, 567–599, https://doi.org/10.1146/annurev-marine-120308-081003, 2011.
Li, D., Luo, R., Liu, C. M., Leung, C. M., Ting, H. F., Sadakane, K., Yamashita, H., and Lam, T. W.: MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices, Methods, 102, 3–11, https://doi.org/10.1016/j.ymeth.2016.02.020, 2016.
Li, H., Wang, H., Wang, H. T., Xin, P. Y., Xu, X., Ma, Y., Liu, W. P., Teng, C. Y., Jiang, C., Lou, L. P., Arnold, W., Cralle, L., Zhu, Y. G., Chu, J. F., Gilbert, J. A., and Zhang, Z. J.: The chemodiversity of paddy soil dissolved organic matter correlates with microbial community at continental scales, Microbiome, 6, 187, https://doi.org/10.1186/s40168-018-0561-x, 2018.
Li, Y. and Cai, Y.: Progress in the study of mercury methylation and demethylation in aquatic environments, Sci. Bull., 58, 177–185, https://doi.org/10.1007/s11434-012-5416-4, 2012.
Liaw, A. and Wiener, M. C.: Classification and Regression by randomForest, R News, 2, 18–22, CRAN, https://cran.r-project.org/package=randomForest (last access: 24 April 2024), 2002.
Liu, J., Lu, B., Poulain, A. J., Zhang, R., Zhang, T., Feng, X., and Meng, B.: The underappreciated role of natural organic matter bond Hg(II) and nanoparticulate HgS as substrates for methylation in paddy soils across a Hg concentration gradient, Environ. Pollut., 292, 118321, https://doi.org/10.1016/j.envpol.2021.118321, 2022.
Liu, J., Chen, J., Poulain, A. J., Pu, Q., Hao, Z., Meng, B., and Feng, X.: Mercury and Sulfur Redox Cycling Affect Methylmercury Levels in Rice Paddy Soils across a Contamination Gradient, Environ. Sci. Technol., 57, 8149–8160, https://doi.org/10.1021/acs.est.3c02676, 2023.
Liu, Y., Johs, A., Li, B., Lu, X., Hu, H., Sun, D. H., He, J. Z., and Gu, B.: Unraveling Microbial Communities Associated with Methylmercury Production in Paddy Soils, Environ. Sci. Technol., 52, 13110–13118, https://doi.org/10.1021/acs.est.8b03052, 2018.
Martin, M.: Cutadapt removes adapter sequences from high-throughput sequencing reads, EMBnet J., 17, 10–12, https://doi.org/10.14806/ej.17.1.200, 2011.
Meng, B., Feng, X., Qiu, G., Liang, P., Li, P., Chen, C., and Shang, L.: The process of methylmercury accumulation in rice (Oryza sativa L.), Environ. Sci. Technol., 45, 2711–2717, https://doi.org/10.1021/es103384v, 2011.
Muthayya, S., Sugimoto, J. D., Montgomery, S., and Maberly, G. F.: An overview of global rice production, supply, trade, and consumption, Ann. New York Acad. Sci., 1324, 7–14, https://doi.org/10.1111/nyas.12540, 2014.
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O'Hara, R. B., Simpson, G. L., Solymos, P., Steven, M. H. H., and Wagner, H.: Vegan: community ecology package, R package version 2.4-4, http://CRAN.R-project.org/package=vegan (last access: 24 April 2024), 2017.
Oloo, F. O., Valverde, A., Quiroga, M. V., Vikram, S., Cowan, D. A., and Mataloni, G.: Habitat heterogeneity and connectivity shape microbial communities in South American peatlands, Sci. Rep., 6, 25712, https://doi.org/10.1038/srep25712, 2016.
Oulhote, Y., Debes, F., Vestergaard, S., Weihe, P., and Grandjean, P.: Aerobic Fitness and Neurocognitive Function Scores in Young Faroese Adults and Potential Modification by Prenatal Methylmercury Exposure, Environ. Health Persp., 125, 677–683, https://doi.org/10.1289/EHP274, 2017.
Pankratov, T. A. and Dedysh, S. N.: Granulicella paludicola gen. nov., sp. nov., Granulicella pectinivorans sp. nov., Granulicella aggregans sp. nov. and Granulicella rosea sp. nov., acidophilic, polymer-degrading acidobacteria from Sphagnum peat bogs, Int. J. Syst. Evol. Microbiol., 60, 2951–2959, 2010.
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W.: CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes, Genome Res., 25, 1043–1055, https://doi.org/10.1101/gr.186072.114, 2015.
Parks, D. H., Chuvochina, M., Rinke, C., Mussig, A. J., Chaumeil, P. A., and Hugenholtz, P.: GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy, Nucleic Acids Res., 50, D785–D794, https://doi.org/10.1093/nar/gkab776, 2022.
Parks, J. M., Johs, A., Podar, M., Bridou, R., Hurt, R. A., Smith, S. D., Tomanicek, S. J., Qian, Y., Brown, S. D., Brandt, C. C., Palumbo, A. V., Smith, J. C., Wall, J. D., Elias, D. A., and Liang, L.: The Genetic Basis for Bacterial Mercury Methylation, Science, 339, 1332–1335, https://doi.org/10.1126/science.1230667, 2013.
Peterson, B. D., Krabbenhoft, D. P., McMahon, K. D., Ogorek, J. M., Tate, M. T., Orem, W. H., and Poulin, B. A.: Environmental formation of methylmercury is controlled by synergy of inorganic mercury bioavailability and microbial mercury-methylation capacity, Environ. Microbiol., 25, 1409–1423, https://doi.org/10.1111/1462-2920.16364, 2023.
Podar, M., Gilmour, C. C., Brandt, C., Soren, A. B., Brown, S. D., Crable, B. R., Palumbo, A., Somenahally, A., and Elias, D. A.: Global prevalence and distribution of genes and microorganisms involved in mercury methylation, Sci. Adv., 1, e1500675, https://doi.org/10.1126/sciadv.1500675, 2015.
Qi, Y., Fu, W., Tian, J., Luo, C., Shan, S., Sun, S., Ren, P., Zhang, H., Liu, J., Zhang, X., and Wang, X.: Dissolved black carbon is not likely a significant refractory organic carbon pool in rivers and oceans, Nat. Commun., 11, 5051, https://doi.org/10.1038/s41467-020-18808-8, 2020.
Revelle, W.: psych: Procedures for Psychological, Psychometric, and Personality Research, Northwestern University, Evanston, Illinois, USA, https://CRAN.R-project.org/package=psych (last access: 5 June 2024), 2023.
Roman, H. A., Walsh, T. L., Coull, B., Dewailly, É., Guallar, E., Hattis, D. B., Mariën, K., Schwartz, J. D., Stern, A. H., Virtanen, J. K., and Rice, G. E.: Evaluation of the Cardiovascular Effects of Methylmercury Exposures: Current Evidence Supports Development of a Dose–Response Function for Regulatory Benefits Analysis, Environ. Health Persp., 119, 607–614, https://doi.org/10.1289/ehp.1003012, 2011.
Sakai, S., Conrad, R., Liesack, W., and Imachi, H.: Methanocella arvoryzae sp. nov., a hydrogenotrophic methanogen isolated from rice field soil, Int. J. Syst. Evol. Micr., 60, 2918–2923, https://doi.org/10.1099/ijs.0.020883-0, 2010.
Schartup, A. T., Ndu, U., Balcom, P. H., Mason, R. P., and Sunderland, E. M.: Contrasting effects of marine and terrestrially derived dissolved organic matter on mercury speciation and bioavailability in seawater, Environ. Sci. Technol., 49, 5965–5972, https://doi.org/10.1021/es506274x, 2015.
Schartup, A. T., Thackray, C. P., Qureshi, A., Dassuncao, C., Gillespie, K. M., Hanke, A. R., and Sunderland, E. M.: Climate change and overfishing increase neurotoxicant in marine predators, Nature, 572, 648–650, https://doi.org/10.1038/s41586-019-1468-9, 2019.
Schöne, C., Poehlein, A., Jehmlich, N., Adlung, N., Daniel, R., von Bergen, M., Scheller, S., and Rother, M.: Deconstructing Methanosarcina acetivorans into an acetogenic archaeon, P. Natl. Acad. Sci. USA, 119, e2113853119, https://doi.org/10.1073/pnas.2113853119, 2022.
Skyllberg, U., Bloom, P. R., Qian, J., Lin, C.-M., and Bleam, W. F.: Complexation of mercury(II) in soil organic matter: EXAFS evidence for linear two-coordination with reduced sulfur groups, Environ. Sci. Technol., 40, 4174–4180, https://doi.org/10.1021/es0600577, 2006.
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., Djahanschiri, B., Zeller, G., Mende, D. R., Alberti, A., Cornejo-Castillo, F. M., Costea, P. I., Cruaud, C., d'Ovidio, F., Engelen, S., Ferrera, I., Gasol, J. M., Guidi, L., Hildebrand, F., Kokoszka, F., Lepoivre, C., Lima-Mendez, G., Poulain, J., Poulos, B. T., Royo-Llonch, M., Sarmento, H., Vieira-Silva, S., Dimier, C., Picheral, M., Searson, S., Kandels-Lewis, S., Bowler, C., Vargas, C. d., Gorsky, G., Grimsley, N. H., Hingamp, P., Iudicone, D., Jaillon, O., Not, F., Ogata, H., Pesant, S., Speich, S., Stemmann, L., Sullivan, M. B., Weissenbach, J., Wincker, P., Karsenti, E., Raes, J., Acinas, S. G., and Bork, P.: Structure and function of the global ocean microbiome, Science, 348, 1261359, https://doi.org/10.1126/science.1261359, 2015.
Tao, S., Fang, J., Zhao, X., Zhao, S., Shen, H., Hu, H., Tang, Z., Wang, Z., and Guo, Q.: Rapid loss of lakes on the Mongolian Plateau, P. Natl. Acad. Sci. USA, 112, 2281–2286, https://doi.org/10.1073/pnas.1411748112, 2015.
Ullrich, S. M., Tanton, T. W., and Abdrashitova, S. A.: Mercury in the Aquatic Environment: A Review of Factors Affecting Methylation, Crit. Rev. Env. Sci. Tec., 31, 241–293, https://doi.org/10.1080/20016491089226, 2001.
Uritskiy, G., DiRuggiero, J., and Taylor, J.: MetaWRAP – a flexible pipeline for genome-resolved metagenomic data analysis, Microbiome, 6, 158, https://doi.org/10.1186/s40168-018-0541-1, 2018.
Wang, L., Wang, L.-A., Zhan, X., Huang, Y., Wang, J., and Wang, X.: Response mechanism of microbial community to the environmental stress caused by the different mercury concentration in soils, Ecotox. Environ. Safe, 188, 109906, https://doi.org/10.1016/j.ecoenv.2019.109906, 2020.
Wickham, H. (Eds.): ggplot2 – Elegant Graphics for Data Analysis, Springer-Verlag, New York, https://doi.org/10.1007/978-0-387-98141-3, 2009.
Xenopoulos, M. A., Barnes, R. T., Boodoo, K. S., Butman, D. E., Catalán, N., D'Amario, S. C., Fasching, C., Kothawala, D., Pisani, O., Solomon, C. T., Spencer, R. G. M., Williams, C. J., and Wilson, H. F.: How humans alter dissolved organic matter composition in freshwater: relevance for the Earth's biogeochemistry, Biogeochemistry, 154, 323–348, https://doi.org/10.1007/s10533-021-00753-3, 2021.
Xun, W., Liu, Y., Li, W., Ren, Y., Xiong, W., Xu, Z., Zhang, N., Miao, Y., Shen, Q., and Zhang, R.: Specialized metabolic functions of keystone taxa sustain soil microbiome stability, Microbiome, 9, 35, https://doi.org/10.1186/s40168-020-00985-9, 2021.
Yin, Y., Li, Y., Ma, X., Liu, J., and Jiang, G.: Role of Natural Organic Matter in the Biogeochemical Cycle of Mercury : Binding and Molecular Transformation, Prog. Chem., 25, 2169–2177, https://doi.org/10.1016/j.scitotenv.2021.152047, 2013
Zhang, H., Feng, X., Larssen, T., Qiu, G., and Vogt, R. D.: In inland China, rice, rather than fish, is the major pathway for methylmercury exposure, Environ. Health Persp., 118, 1183–1188, 2010.
Zhang, R., Aris-Brosou, S., Storck, V., Liu, J., Abdelhafiz, M. A., Feng, X., Meng, B., and Poulain, A. J.: Mining-impacted rice paddies select for Archaeal methylators and reveal a putative (Archaeal) regulator of mercury methylation, ISME Commun., 3, 74, https://doi.org/10.1038/s43705-023-00277-x, 2023.
Zhao, L., Chen, H., Lu, X., Lin, H., Christensen, G. A., Pierce, E. M., and Gu, B.: Contrasting Effects of Dissolved Organic Matter on Mercury Methylation by Geobacter sulfurreducens PCA and Desulfovibrio desulfuricans ND132, Environ. Sci. Technol., 51, 10468–10475, https://doi.org/10.1021/acs.est.7b02518, 2017.
Zhong, H., Tang, W., Li, Z., Sonne, C., Lam, S. S., Zhang, X., Kwon, S. Y., Rinklebe, J., Nunes, L. M., Yu, R. Q., Gu, B., Hintelmann, H., Tsui, M. T. K., Zhao, J., Zhou, X. Q., Wu, M., Liu, B., Hao, Y., Chen, L., Zhang, B., Tan, W., Zhang, X. X., Ren, H., and Liu, Y. R.: Soil Geobacteraceae are the key predictors of neurotoxic methylmercury bioaccumulation in rice, Nat. Food, 5, 301–311, 2024.