the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Mar 2025
| 31 Mar 2025
Stable iron isotope signals indicate a “pseudo-abiotic” process driving deep iron release in methanic sediments
Michael Staubwasser
Simone A. Kasemann
Anette Meixner
David A. Aromokeye
Michael W. Friedrich
Sabine Kasten
The low δ56Fe values of dissolved iron liberated by microbial iron reduction are characteristic of many shallow subsurface sediments and – if not significantly changed within the oxic sediment layer – the related benthic Fe fluxes into the water column. Here, we decipher whether stable Fe isotope signatures in pore water and the respective solid-phase sediment samples are also useful for unraveling the processes driving Fe liberation in deeper methanic sediments. We investigated the fine-grained deposits of the Helgoland mud area, North Sea, where Fe reduction in the methanic subsurface sediments was previously suggested to be coupled to methanogenic fermentation of organic matter and anaerobic methane oxidation. In the evaluated subsurface sediments, a combination of iron isotope geochemistry with reactive transport modeling for the deeper methanic sediments hints at a combination of processes affecting the stable isotope composition of dissolved iron. However, the dominant process releasing Fe at depth does not seem to lead to notable iron isotope fraction. Under the assumption that iron reducing microbes generally prefer isotopically light iron, the deep Fe reduction in this setting appears to be “pseudo-abiotic”: if fermentation is the main reason for Fe release at depth, the fermenting bacteria transfer electrons directly or indirectly to Fe(III), but our data do not indicate notable related isotopic fractionation. Our findings strongly contribute to the debate on the pathway for deep Fe2+ release by showing that the main underlying process is mechanistically different to the microbial Fe reduction dominating in the shallow sediments and encourages future studies to focus on the fermentative degradation of organic matter as a source of dissolved iron in methanic sediments.
- Article
(3731 KB) - Full-text XML
-
Supplement
(49483 KB) - BibTeX
- EndNote




Iron reduction in coastal and marine sediments plays an important role in the degradation of organic matter, the transformation and cycling of carbon species, and benthic nutrient release into the water column (e.g., Baloza et al., 2022; LaRowe and Van Capellen, 2011; Lovley and Phillips, 1986; Thamdrup et al., 1994; Zhou et al., 2023, 2024). Multiple studies, e.g., Henkel et al. (2016, 2018), Johnson and Beard (2005), Severmann et al. (2006), and Staubwasser et al. (2006), have shown that the difference in the isotopic composition of solid Fe(III) and dissolved Fe(II) in shallow marine sediments is similar to the fractionation related to dissimilatory iron reduction (DIR): based on pure-culture studies, dissimilatory iron-reducing microorganisms (e.g., Shewanella spp.) favor the light iron isotope 54Fe, which is therefore preferentially released into pore water while the ferric substrate becomes isotopically heavier (e.g., Beard et al., 1999, 2003a; Johnson et al., 2004, 2005). Part of the microbially liberated (isotopically light) Fe(II) is adsorbed onto the oxide surface and in isotopic exchange with the heavier reactive Fe(III) layer of the oxide. The resulting fractionation (combining DIR and the electron and atom exchange) Δ56FeFe(II)diss−Fe(III) is up to −3 ‰ (e.g., Crosby et al., 2005, 2007). Iron isotopes, expressed as δ56Fe (‰), are thus considered a tool for assessing the role of microbial iron reduction (MIR) for the mineralization of organic matter and for tracing benthic iron fluxes into the water column (e.g., Conway and John, 2014; Homoky et al., 2009; John et al., 2012; Severmann et al., 2006, 2010; Sieber et al., 2021). Here, we aim to evaluate whether pore-water and solid-phase Fe isotope signatures are also useful for unraveling the processes driving Fe reduction in deeper sediments below the sulfate–methane transition (SMT) that is frequently observed in freshwater, brackish, and marine depositional environments (e.g., Egger et al., 2017; Hensen et al., 2003; Kasten et al., 1998; März et al., 2008; Oni et al., 2015a; Riedinger et al., 2005, 2010, 2014; Segarra et al., 2013; Sivan et al., 2011; Wersin et al., 1991). The processes responsible have not been entirely understood so far. Most of the sites at which this “deep Fe reduction” occurs are in high-deposition areas characterized by a rapid transition of Fe-(oxyhydr)oxides through the upper zone of Fe reduction and the following sulfidic interval into methanic, non-sulfidic sediments (e.g., Aromokeye et al., 2020, 2021; Oni et al., 2015a; Riedinger et al., 2005). Dissolved Fe2+ concentrations typically increase below the sulfidic interval surrounding the SMT and may reach several hundreds of micromolar, often exceeding Fe concentrations in the upper ferruginous zone close to the sediment surface (e.g., Riedinger et al., 2005, 2014, 2017).
There are a variety of possible biotic and abiotic pathways for deep Fe reduction. Biotic pathways include continuing DIR by use of organic or inorganic electron donors (e.g., Lovley, 1991; Lovley et al., 1989; Roden and Lovley, 1993), organoclastic fermentative Fe reduction (e.g., Lehours et al., 2010; Lovley and Phillips, 1986), Fe reduction coupled to ammonium oxidation (Bao and Li, 2017), and Fe-coupled anaerobic oxidation of methane (Fe-AOM) (e.g., Aromokeye et al., 2020; Beal et al., 2009; Riedinger et al., 2014; Sivan et al., 2011). It was furthermore discussed whether Fe2+ release can also be linked to iron oxide reduction by methanogens that can perform Fe-AOM (Yu et al., 2022) or switch between methane generation and Fe reduction (Eliani-Russak et al., 2023; Sivan et al., 2016). In contrast, Fe reduction and potentially also Fe2+ liberation can occur (largely) abiotically by reactions with inorganic compounds such as FeS or FeS2 (Bottrell et al., 2010; Mortimer et al., 2011) and hydrogen sulfide (sulfide oxidation by reduction of Fe(III), e.g., Canfield, 1989; Holmkvist et al., 2011; Pyzik and Sommer, 1981; Riedinger et al., 2010; Thamdrup et al., 1993) as well as by reactions with organic molecules (e.g., oxalate), which themselves might be produced by microbial activity (e.g., Burdige, 1993; Ionescu et al., 2015, and references therein). Recently, Aromokeye et al. (2021) suggested that Fe reduction in methanic sediments of the North Sea occurs concomitantly with the use of crystalline Fe-oxides as conduits for interspecies electron transfer between fermentative bacteria and methanogens (methanogenic benzoate fermentation). The mechanistic details of this process are still to be fully understood. Clearly, abiotic and biotic reactions of Fe in marine sediments are closely interrelated with each other. Moreover, all of them are directly or indirectly linked to the biogeochemical cycling of C and S. In order to fully assess these interlinks and in particular to determine their relevance for methane generation and/or consumption, we require a better understanding of deep Fe reduction pathways in natural settings and their dependence on environmental conditions. Differentiation between abiotic and biotic Fe reaction pathways – in particular in methanic environments – would furthermore be of interest for studying life in extreme environments such as the sedimentary deep biosphere, where the availability of degradable organic matter and electron acceptors yielding high standard free energies is often strongly limited (e.g., Heuer et al., 2017; D'Hondt et al., 2004). Here, we investigate whether combined pore-water and solid-phase stable Fe isotope signatures can be used to differentiate between biotic and abiotic Fe reduction pathways in methanic sediments. A similar approach for assessing the dominance of Fe–S reactions over MIR and vice versa using Fe isotopes was successfully applied in shallow sediments of the continental margin off California (Severmann et al., 2006). We also specifically investigate the Fe isotopic signals of crystalline Fe-oxides, including magnetite, because for the site investigated here, these minerals were found to stimulate deep Fe release based on their conductivity (Aromokeye et al., 2021).
To our knowledge, there have only been two studies so far focusing on Fe reduction in methanic sediments that also include pore-water Fe isotope data: Sivan et al. (2011) proposed the occurrence of Fe-AOM in sediments of Lake Kinneret (Israel) and showed a light isotopic composition of pore-water Fe2+ (∼ −2 ‰) in the respective interval. A recent study on very old and compacted sediments of the Nankai Trough off Japan (IODP site C0023) showed that extremely negative pore-water δ56Fe values of up to −5.9 ‰ are most likely derived from a combination of MIR and Rayleigh fractionation, where 56Fe2+ is preferentially adsorbed onto mineral surfaces (Köster et al., 2023).
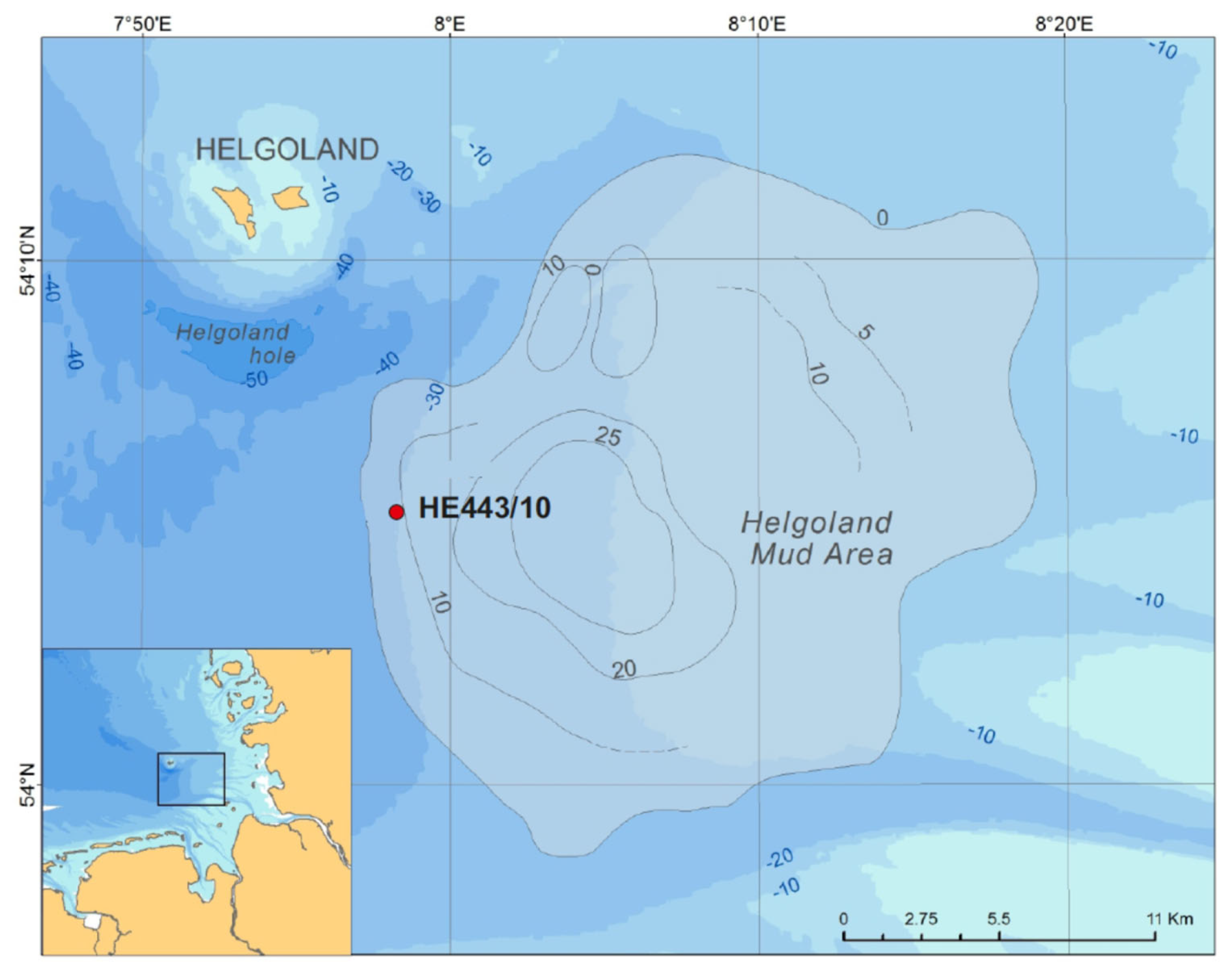
Figure 1Position of cores collected during RV Heincke expedition HE443. Contour lines indicate the thickness of the mud in meters after Hebbeln et al. (2003).
Despite the lack of data concerning Fe isotope fractionation during biotic reduction pathways other than DIR, we assume that microbially mediated Fe liberation in methanic sediments similarly results in a preferential release of 54Fe2+ and, thus, shifts pore-water δ56Fe towards negative values. Furthermore, we hypothesize that the microbial utilization of a specific Fe-(oxyhydr)oxide pool results in relative enrichment of 56Fe in the remaining substrate, enhancement which is detectable by a combination of sequential solid-phase Fe extractions and δ56Fe analyses after Staubwasser et al. (2006) and Henkel et al. (2016, 2018). We present a compilation of inorganic geochemical data including pore-water and bulk solid-phase geochemistry, iron monosulfide and pyrite extractions, and δ56Fe in pore water and reactive Fe pools in order to assess sources and sinks of dissolved iron in the methanic sediments of the Helgoland mud area (HMA, German Bight, North Sea). We exemplarily tested if the δ56Fe data, in combination with geochemical transport-reaction modeling and the information from previous microbiological studies, can be used to trace and explain the contributions of Fe reduction processes at different depths in the sediment column and to discriminate abiotic from biotic reduction pathways.
2.1 Study area
The Helgoland mud area (HMA, Fig. 1) is a halotectonic depression in the German Bight filled with Holocene mud that is mostly discharged by the rivers Elbe and Weser (e.g., Hertweck, 1983; Irion et al., 1987). It is one of the few depocenters of fine-grained and organic-carbon-rich sediments in the German Bight, extends over ∼ 500 km2, and has an average water depth of 20 m (Hebbeln et al., 2003). Irion et al. (1987) explained that between 10 000 and 8000 BP the Elbe estuary was located at the present position of the HMA and that the old glacial relief formed a barrier towards the north that allowed the mud deposition in a comparatively protected bight. According to Irion et al. (1987), this protective barrier was destroyed at about 3000 to 2000 BP due to wave and tide activities. High sedimentation rates of more than 13 mm yr−1 characterized the HMA between ∼ 1250–700 BP and were attributed to the disintegration of the island of Helgoland in the Middle Ages (Hebbeln et al., 2003). The reduction in sedimentation rates to < 3 mm yr−1 after ∼ 700 BP was linked to a slowdown of the disintegration of the island and/or a change in the deposition location (Hebbeln et al., 2003). The high sedimentation rates that prevailed in the region of the HMA before 700 BP led to a fast burial and good preservation of reactive Fe-(oxyhydr)oxides, which is a prerequisite of deep Fe reduction.
Previous investigations in the western have HMA shown a deep Fe release from sediments below the sulfidic zone. The studies by Oni et al. (2015a) and Aromokeye et al. (2020, 2021) demonstrated that the Fe release is directly or indirectly related to microbial activity. Oni et al. (2015a) found a correlation of dissolved iron (Fediss) concentrations with the abundance of Atribacterota (formerly known as candidate phylum JS1), methanogens, and Methanohalobium/anaerobic methanotrophic archaea-3 (ANME-3)-related archaea. It was therefore suggested that these microbes could be involved in deep Fe reduction, possibly via simultaneous occurrence of methanogenesis and Fe-mediated AOM. Aromokeye et al. (2020) performed an incubation experiment on sediment material from the same site as ours (parallel core, site HE443/10) and could detect Fe-AOM in slurries from the methanic zone, in particular in long-term incubations (250 d) amended with synthetic Fe-oxides. Dissolved Fe was, however, also released from sediments of the methanic zone when those were incubated with N2 in the headspace (so unrelated to methane oxidation), irrespective of synthetic Fe-oxide addition. The authors therefore concluded that there are additional processes for deep Fediss generation that are directly linked to organic matter (OM) degradation. One of these pathways was later suggested to be coupled to the fermentation of complex organic matter (Aromokeye et al., 2021).
2.2 Sediment and pore-water sampling
The data shown here are derived from a multiple-corer (MUC) deployment and a gravity core (GC) collected during RV Heincke cruise HE443 in April 2015 (MUC: 54°05.14′ N, 7°58.15′ E; GC: 54°05.19′ N, 7°58.21′ E; water depth 30 m, Fig. 1). The GC, site HE443/10-3, had a length of 488 cm. As surface sediment is usually lost during GC coring, the data of the MUC, site HE443/10-2, were used to fit GC pore-water data and calculate actual sediment depths for GC samples.
Directly after coring, the GC was cut into 1 m segments. Samples for methane (CH4) analysis (3 mL of sediment) were taken immediately at the segment ends and stored in headspace vials that were pre-filled with 20 mL of a saturated NaCl solution containing NaN3. The vials were sealed and stored at 4 °C until analysis. The GC segments were further sampled through small windows cut into the liner. First, additional CH4 samples were collected. Then, pore water was extracted at 20 cm intervals using Rhizon samplers with an average pore size of 0.1 µm (Seeberg-Elverfeldt et al., 2005). The first ∼ 1.5 mL of collected pore water was discarded. Afterwards, 1.5 mL was collected and mixed with zinc acetate (ZnAc) for onshore sulfide measurements, 2 mL was stored in glass vials without headspace for dissolved inorganic carbon (DIC) analysis, 400 µL was diluted 1:10 with a NaCl solution for ammonium (NH4) and phosphate () analyses, and ∼ 1 mL was stored for onshore sulfate () and chloride (Cl−) measurements. After collection of pore water for the abovementioned parameters, new syringes pre-filled with 50 µL of concentrated double-distilled HNO3 were attached to the Rhizon samplers and another 1–2 mL was collected for cation analysis. Pore-water aliquots were all stored at 4 °C, except for nutrient samples, which were stored at −20 °C. To maximize the sample volume, pore water for δ56Fe analyses was collected in between the depths sampled for all other pore-water parameters, also at 20 cm intervals. However, we only processed every second to third sample and based the sample selection on the Fediss profile shape with a higher resolution close to reaction fronts and a lower resolution where Fediss shows a rather linear gradient. Those samples were collected in pre-cleaned PP vials: for 1 d, 3 % ELMA 70 (an alkaline detergent); for 1 d, deionized water; for 7 d, 0.3 M HCl; and for 3 d, ultra-pure water. All δ56Fe samples were acidified with double-distilled HCl. Solid-phase samples were collected using cutoff syringes which were then tightly sealed and stored at −20 °C in Ar-filled gastight glass containers. The GC was cut open 6 months after coring, and X-radiographs were produced at the Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research (AWI) in Bremerhaven.
We collected three parallel MUCs during one deployment at the same location that was cored with the GC: one for pore-water δ56Fe sampling, one for solid-phase analyses, and one for all other parameters mentioned above for the GC except for CH4. Pore-water sampling was conducted as described above but at 1 cm intervals down to a depth of 10 cm and every 2 cm further below. Solid-phase samples were gained by slicing the MUC core at 1–2 cm intervals. These samples were all treated as previously described.
Both the MUC and the GC core, HE443/10-2 and HE443/10-3, consisted of dark to very dark grey mud. The GC that was cut open onshore showed bioturbation structures over the whole core length. Only at the following intervals (core depths) was sediment lamination still largely intact: 20 to 27 cm, 121 to 135 cm, 190 to 195 cm, 238 to 264 cm, 273 to 278 cm, 311 to 326 cm, 340 to 388 cm, and 441 to 447 cm. Layers and lenses of silty material were present at 60, 275, 350 to 360, and 370 to 380 cm. X-radiographs of the core are accessible in the PANGAEA database (Henkel et al., 2024a).
2.3 Pore-water analyses
The following pore-water analyses were conducted at the AWI: DIC, NH4, and PO4 were measured directly after the cruise using a QuAATro SEAL Analytical nutrient analyzer. The methods used were as described in the user handbook (Q-067-05, Q-080-06, Q-064-05) and are based on Stoll et al. (2001) (for DIC), Kerouel and Aminot (1997) (for NH4), and the complex formation of with ammonium molybdate, respectively. The pH value was measured in sampled pore water using a pH electrode and a WTW pH meter. Cation concentrations (Fe, Mn, Ca, Mg) were determined by inductively coupled plasma–optical emission spectrometry (ICP-OES, IRIS Intrepid II). Dissolved sulfide () was analyzed using the methylene blue method (Cline, 1969). Sulfate and chloride were measured in 1:50 dilution using a Metrohm 761 Compact IC ion chromatograph. Headspace gas for CH4 analysis was injected into a Thermo Finnigan TRACE GC equipped with a packed column and an integrated flame ionization detector (FID). Methane concentrations were calculated under consideration of an average porosity of 0.7, determined based on the water content of sediment samples and an estimated average grain density of 2.6 g cm−3.
Pore-water processing for δ56Fe analysis was conducted at the University of Cologne: Fe was first pre-concentrated and extracted from the salt matrix using the NTA Superflow resin as described by Henkel et al. (2016, 2018). Subsequently, samples were further purified by anion exchange chromatography using 150 µL Dowex 1X8 200-400 resin. All columns and vials used during the processing were pre-cleaned with ELMA 70 and diluted HCl as described above for the PP sampling vials. The purified Fe samples were matched to a concentration of 0.2 ppm and introduced into a Thermo Finnigan Neptune multicollector inductively coupled plasma–mass spectrometry (ICP-MS) instrument equipped with an Aridus desolvating nebulizer system at the Steinmann Institute in Bonn. We applied the standard-sample bracketing method with IRMM-014 (e.g., Schoenberg and von Blanckenburg, 2005). All 54Fe data were Cr-corrected based on measurements of 53Cr. In addition, all data were blank-corrected and samples were analyzed in random order. Data are reported as δ56Fe [‰] = . Details concerning the instrumental setup can be found in Henkel et al. (2016, 2018). We monitored the measurement trueness of the isotopic analyses by use of the reference material JM, a solution produced from an Fe wire supplied by Johnson Matthey. A JM was measured after each set of six samples. The measured value was 0.49 ± 0.26 ‰ (n=15, 2 SD) and overlapped within uncertainty with previously published values (0.42 ± 0.05 ‰, Schoenberg and von Blanckenburg, 2005; 0.46 ± 0.20 ‰, Walczyk and von Blanckenburg, 2005; 0.35 ± 0.14 ‰, Weyer and Schwieters, 2003). The uncertainty in a single measurement (2 SD of one block of 20 measurement cycles) was between 0.07 and 0.14 ‰. Processing and analysis of two duplicate samples resulted in δ56Fe values within the uncertainties (2 SD) of the respective single measurements. The trueness of the entire sample processing procedure was controlled by processing of blanks, the isotope standard IRMM-014, and Fe standards (Certipur®) in different concentrations. Blanks yielded Fe concentrations of 3 ppb, a level which was at most 4 % of the Fe concentration in the pore-water samples. The processing of Certipur® Fe standards showed a recovery of > 85 % of Fe. Pore-water δ56Fe values were analyzed using a Keeling plot, which is traditionally used for carbon isotope mixing (Keeling, 1958; Pataki et al., 2003). Details are given in Sect. 4.2.
2.4 Solid-phase analyses
The bulk elemental composition of the sediment was determined by total digestion of about 50 mg of freeze-dried and ground sediment in a mixture of concentrated acids (3 mL HCl, 2 mL HNO3, and 0.5 mL HF). The digestion was carried out in a CEM MARSXpress microwave system at AWI. After evaporation of the acids, the residue was dissolved in 1 M HNO3 and measured by ICP-OES. Recoveries of processed NIST SRM 2702 reference material (n=5, uncertainty given as 2 SD) were 100.2 ± 0.8 % for Fe, 98.1 ± 2.4 % for Mn, 101.5 ± 2 % for Ca, and 93.8 ± 2.8 % for Al. Total Fe contents were published by Aromokeye et al. (2020) and are available in PANGAEA (Aromokeye et al., 2018a).
Sequential Fe extractions were performed after Poulton and Canfield (2005): ∼ 50 mg of dry sediment was suspended in 5 mL of (a) MgCl2 for adsorbed Fe, (b) Na-acetate for Fe-carbonates and surface-reduced Fe(II), (c) hydroxylamine–HCl for easily reducible Fe-oxides (ferrihydrite, lepidocrocite), (d) Na-dithionite/Na-citrate for reducible Fe-oxides (goethite and hematite), and (e) ammonium oxalate/oxalic acid for magnetite. In comparison to the method by Poulton and Canfield (2005), we used a lower concentration of citrate for the dithionite extraction since citrate hinders Fe precipitation during subsequent sample purification for δ56Fe analysis (Henkel et al., 2016). Instead, we performed the extractions under anoxic conditions. Aliquots of all extracts were analyzed by ICP-OES. A separate aliquot of 2 mL of each extract was processed for δ56Fe analysis following the protocol by Henkel et al. (2016). Henkel et al. (2016) demonstrated that the processing of samples does not lead to iron fractionation. The purified Fe samples were matched to a concentration of 0.5 ppm and were analyzed using the Thermo Fisher Scientific Neptune Plus MC ICP-MS instrument of the Isotope Geochemistry Group at MARUM – Center for Marine Environmental Sciences, University of Bremen. The MC ICP-MS instrument was equipped with an SSI dual cyclonic spray chamber, a low-flow 50 µL PFA nebulizer, and a Ni skimmer cone (X-type). Samples were measured using the standard-sample bracketing with certified reference material IRMM-014. All 54Fe data were Cr-corrected based on measurements of 52Cr. In addition, all data were blank-corrected and samples were analyzed in random order. The standard JM (see above) was analyzed after each block of three samples. Samples bracketed by JMs that did not fall into the target range of 0.42 ± 0.05 ‰ were repeatedly measured. The repeatability precision resulting from up to six replicate sample measurements (not including replicate processing) was in the worst case 0.34 ‰ (2 SD) and in the best case 0.03 ‰ (average 0.11 ‰; see Fig. 5). The intermediate precision of JMs was 0.44 ± 0.15 ‰ (n=151, 2 SD).
Acid volatile sulfide (AVS; mostly iron monosulfides) and chromium reducible sulfide (CRS; mostly pyrite but potentially also elemental sulfur) were extracted at AWI by hot digestion using 6 M HCl and a chromous chloride solution, respectively (Canfield et al., 1986; Praharaj and Fortin, 2003; Wieder et al., 1985). Extracted sulfur was trapped in a silver nitrate solution as Ag2S. After filtration, the dry masses of the precipitates were converted into FeS and FeS2 contents based on stoichiometry. Replicate analysis of an in-house standard (core catcher sediment of GC HE443/077) revealed good reproducibility of the extractions with AVS contents of 0.11 ± 0.01 wt % and pyrite contents of 1.03 ± 0.05 wt % (n=7).
On freeze-dried, powdered, and homogenized sediment samples, the total carbon (TC) contents were determined using a CNS (Elementar vario EL III) analyzer. Total organic carbon (TOC) contents were measured with a carbon–sulfur analyzer (CS-2000, ELTRA) after removal of inorganic carbon with HCl.
2.5 Model setup and parameterization
A reactive transport model was used in order to (1) exemplarily assess if the measured pore-water Fe and δ56Fe profiles at site HE443/10 can be reproduced based on Fe reactions that are known to occur (MIR and Fe sulfide formation) and (2) delineate how sensitive the pore-water Fe and δ56Fe profiles are with respect to different reaction rate constants k and related fractionation factors. To keep this approach as simple and straightforward as possible, we only included the most basic and presumably dominant reactions that are known to affect the dissolved Fe pool and its isotopic composition: organoclastic DIR, reaction of hydrogen sulfide with Fe(III) to Fe sulfide, and the precipitation of Fe sulfides by counter-diffusion of pore water Fe2+ and HS− (Reactions R1–R3, Table S1 in the Supplement).
We are aware that we miss some reactions in the model that might play a role as well, e.g., siderite or vivianite precipitation, Fe-AOM, and reoxidation of sulfide by Fe(III). Furthermore, Reaction (R3) is actually not a single reaction but includes the formation of monosulfide (FeS) and (in a second step) the transformation of FeS into FeS2, where the latter reaction can happen abiotically but can also be driven by microbes (Thiel et al., 2019). Our approach is basically backwards as we check whether we can reproduce the profile shapes of dissolved Fe and the respective δ56Fe values from the deep Fe source to the sink at the sulfidization front sufficiently well by just including these basic reactions or whether we miss a reaction that would be needed to explain the measurements. With regard to siderite and vivianite formation, a calculation with PHREEQC (see Sect. 2.6) in fact indicates oversaturation below the SMT at site HE443/10-3. Nevertheless, we chose to neglect these reactions in the model as the specific contributions are unclear (siderite and vivianite) or respective Fe fractionation factors are unknown (vivianite). We discuss, however, how particularly siderite precipitation could affect our results, e.g., Fe extraction data and the dissolved Fe isotope profile in Sect. 4.1 and 4.2. Since we were primarily interested in the deep Fe reduction, modeling was confined to the sediment interval between 70 cm (sulfide peak) and 450 cm (end of core). We disregarded all the reduction and oxidation processes above the sulfide peak as they are irrelevant for the expression of aqueous δ56Fe below the sulfidic zone. This is because Fe2+ is completely removed within the sulfidic zone due to the reaction with HS−.
The reaction rates were obtained according to the concentrations of the reacting species. For example, Reactions (R2) and (R3) approach zero when HS− is depleted. The following transport-reaction equations for Fe2+ and HS− were used:
where [Fe2+] and [HS−] are the concentrations of dissolved iron and sulfide, t is time, ω is the sedimentation rate, z is the depth below seafloor, and and are the diffusion constants for dissolved iron and sulfide. The applied sedimentation rate (0.16 cm yr−1) is derived from Hebbeln et al. (2003). Diffusion constants for seawater at pore-water temperature (4 °C) are from Boudreau (1996b) ( = 0.0116 m2 yr−1 and = 0.0306 m2 yr−1). A constant porosity (φ) of 0.7 was assumed, and the tortuosity (τ) in Eqs. (1) and (2) was calculated according to Boudreau (1996a) as . Although the OM degradation rate constant k1 decreases with burial depth (Arndt et al., 2013), its variation is low below the SMT under high-burial-rate conditions. For simplicity, the Fe reduction rate coupled with OM degradation is assumed to follow the first-order decay model (E1; see Table S1). The pool of reducible Fe-oxides is set to not be limiting and is based on the data gained from sequentially extracted Fe, and its δ56Fe composition was kept at a constant value of 0.0 ‰ (see Results). Within the sulfidic zone there is no free Fe2+ (assumption: 0.01 µM at the upper boundary), and all Fe2+ released from the abiotic reaction with HS− (sulfide oxidation by reduction of iron (oxyhydr)oxides) is assumed to be immediately converted into pyrite (Reaction R2). Due to the complete turnover of released Fe2+, it is reasonable to assume that there is no related isotopic fractionation (α2 = 1.000). During Fe sulfide formation where Fe2+ and HS− counter-diffuse, we applied the kinetic fractionation factor α3 (), which was set to 0.999, 0.998, and 0.997 to fit the measured data and resulted in dissolved Fe with an isotopic composition δ56Fediss ∼ 0 ‰ compared to more negative values presented below (see Results and Discussion), where Fe concentrations are higher (upward Fe diffusion). In other words, Fe sulfide formation via the reaction of Fe2+ with HS− preferentially incorporates 54Fe (e.g., Butler et al., 2005; Scholz et al., 2014; Severmann et al., 2006). In addition, in different model runs we applied a fractionation factor = 0.998, 0.997, and 0.996, for DIR below 70 cm depth in order to reproduce the measured pore-water δ56Fe profile. These values are in the range of DIR fractionation factors published by Beard et al. (1999, 2003b), Crosby et al. (2007), Johnson et al. (2005), and Severmann et al. (2006). It is important to note that the factors we apply do not resolve all the partial fractionation processes involved but only the fractionation related to the sum of Reactions (R1) and (R3). Our α1 for example reflects the isotopic difference between solid-phase Fe(III) and dissolved Fe(II), but in reality isotopic fractionation happens not only during the microbial Fe reduction and Fe2+ release, but also between adsorbed Fe(II) and a reactive solid Fe(III) and adsorbed Fe(II) and the dissolved Fe(II), respectively (Crosby et al., 2005, 2007). Furthermore, we note that it is a valid approach to use k as a fitting parameter because for biotic reactions, the rate constant depends not only on temperature, but also on the abundance and activity of microbes. Typically, this leads to a very large range of constant values. For example, the rate constant was given as 100 and 14 800 for the same reaction, , in Reed et al. (2011a) and Reed et al. (2011b), respectively. In our study, k2 also depends on the contents of reactive Fe(OH)3 because Reaction (R2) is generally expressed as k[Fe(OH)3][H2S]. k3 combines the fast FeS formation rate constant k[Fe2+][H2S] and slow FeS2 rate constant k[FeS][H2S]. All model parameters and boundary conditions are given in Table S2 in the Supplement.
2.6 Calculation of saturation indices (SIs)
The saturation indices of selected secondary Fe minerals, namely vivianite and siderite, were calculated using the computer program PHREEQC (Parkhurst and Appelo, 2013). The thermodynamic database “phreeqc.dat” was used because it has a relatively wide range of aqueous complexation reactions for 25 chemical elements, including P and Fe. The input files defined for the geochemical calculations in PHREEQC are based on measured DIC; pH; and the aqueous concentrations of Mg2+, , , , HS−, Cl−, Mn2+, and Fe2+. was set to zero as it is already depleted close to the sediment surface. Since the redox potential (Eh) is a mandatory input variable for these types of geochemical calculations but was unavailable, the default values in PHREEQC were used. The effect of Eh on the saturation of vivianite and siderite was insignificant as determined by a sensitivity test. The in situ temperature of the pore water was set to 4 °C. The concentration of Fe2+ within the sulfidic zone was set to 1 µM as the detection limit.
Based on the DIC and profiles of the MUC and GC cores, the core loss during gravity coring was determined to be 16 cm. Core depths of the GC were corrected to sediment depth accordingly.
3.1 Pore-water geochemistry
The pore-water profiles of , HS−, CH4 (only end of core segments), DIC, and dissolved Fe and Mn of GC HE443/10-3 were shown earlier in Aromokeye et al. (2020) and are available in the PANGAEA database (Aromokeye et al., 2018b; core depths instead of sediment depths).
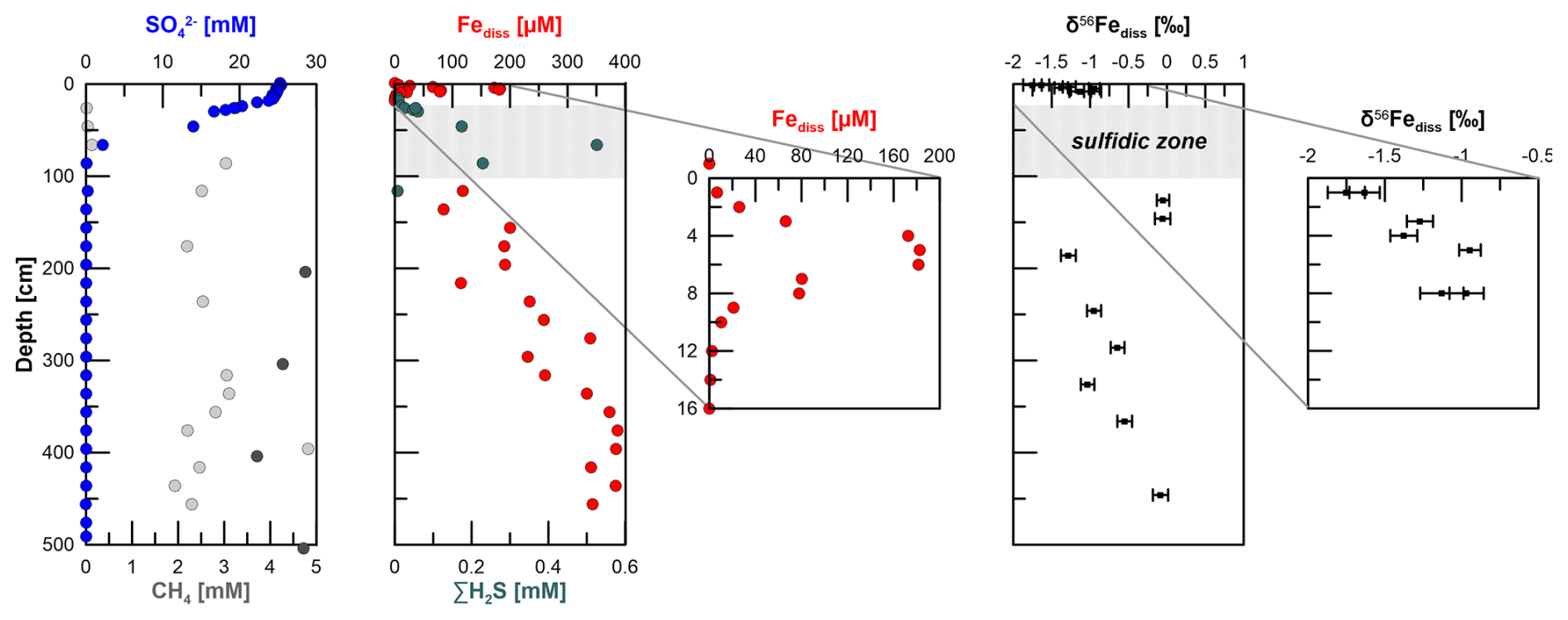
Figure 2Pore-water profiles of and CH4 at station HE443/10. Dark grey dots of the CH4 graph indicate samples from ends of core segments. Those concentrations are more reliable compared to the others, as samples were directly taken during cutting of the core segments. The second and third panels show dissolved Fe and H2S concentrations as well as the stable Fe isotope values of dissolved Fe in the non-sulfidic sediments (uncertainty bars are 2 SD determined for the 20 measurement cycles in one “block” of analysis). Sulfate, methane (only end of core segments), sulfide, and dissolved-iron data of the gravity core were published earlier by Aromokeye et al. (2020). In all plots, the grey-shaded area indicates the sulfidic interval.
The pore-water profiles at site HE443/10 indicate ferruginous conditions at 1–2 cm depth. Dissolved Fe concentrations peak at 5 cm with 180 µM and then decrease towards 10 cm depth, where Fe and dissolved sulfide counter-diffuse (Fig. 2). Based on concurrent analysis of Fe2+ (using the ferrozine method after Stookey, 1970) and dissolved Fe with ICP-OES on several other sites of the same expedition, we are confident that all dissolved Fe at station HE443/10 is in the form of Fe2+. Sulfate shows a kink-shaped profile with a minor decrease from the sediment–water interface to 15 cm depth (25.3 to 24.3 mM) and a steeper gradient further downcore to ∼ 0 mM at 86 cm. Sulfidic conditions prevail between 10 and 100 cm depth. Sulfide concentrations in the pore water peak at ∼ 70 cm depth (0.5 mM), where and CH4 counter-diffuse. Right below the SMT, CH4 concentrations increase to ∼ 3 mM and more. Higher values, ≥ 4 mM, were measured in samples directly taken from ends of core segments during the cutting of the core. At depth, CH4 concentrations do not significantly increase. Below the sulfidic zone, dissolved Fe (Fediss) concentrations gradually increase downcore to 400 µM at 350 cm. The concentrations remain at this level further below. The δ56Fediss profile shows the most negative values at the sediment–water interface (−1.8 ‰) (Fig. 2). The values increase to about −1 ‰ at 8 cm depth. There are no δ56Fediss values for the sulfidic zone due to absence of Fediss. Right below the sulfidic zone, where Fediss concentrations of ∼ 100 µM were detected, δ56Fediss is 0 ‰. As Fediss concentrations increase further downcore, there is a shift to −1.3 ‰ at 186 cm followed by a gradual increase in values towards 0 ‰ at 450 cm, where Fediss peaks.
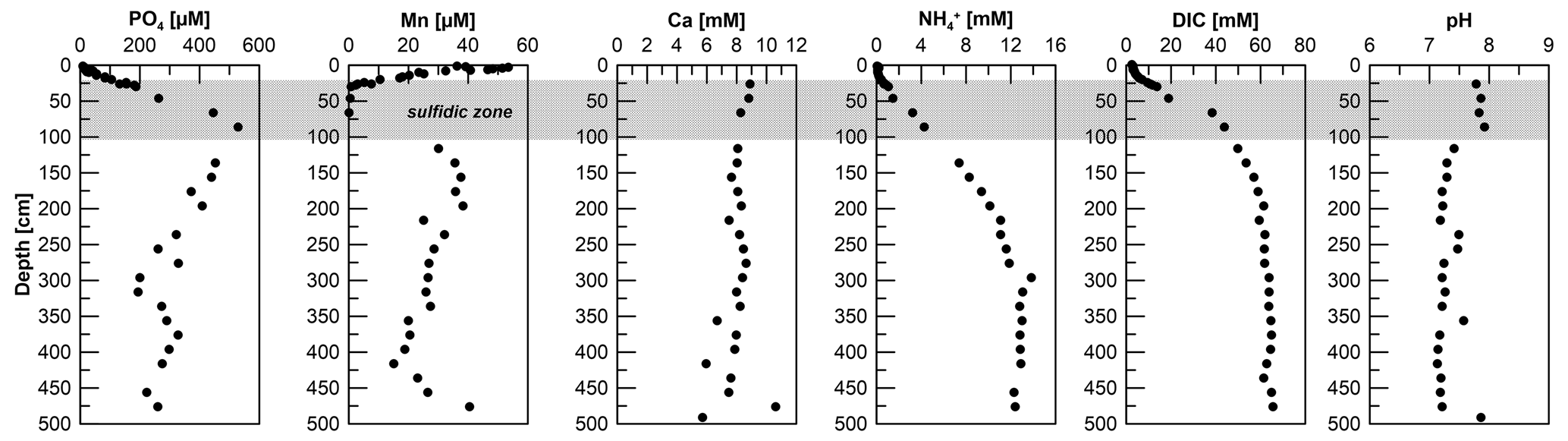
Figure 3Pore-water PO4, Mn, Ca, , DIC, and pH data for site HE443/10. The Mn pore water profile of the gravity core was published earlier by Aromokeye et al. (2020).
Phosphate concentrations show an increase from 9 µM at 1 cm depth to ∼ 530 µM at 90 cm. Concentrations then decrease gradually to ∼ 250 µM at depth (Fig. 3). Phosphorus concentrations measured by ICP-OES of acidified pore-water aliquots (not shown but available under Henkel et al., 2024a) mirror the overall PO4 profile so that we can exclude a drawdown of PO4 at depth as a sampling artifact in Fe-rich pore water from below the SMT. Oxidation of samples easily leads to Fe precipitation and PO4 drawdown due to adsorption. The Mn pore-water profile shows concentrations of ∼ 50 µM at 3 cm sediment depth. Towards the sulfidic zone, concentrations decrease to zero. A second maximum of Mn concentrations (∼ 200 µM) is located at 200 cm. Down to this depth, the Mn profile shape mirrors the Fediss, although concentrations are considerably lower. Unlike Fediss, Mn concentrations then decrease to 15 µM at 415 cm. Towards the end of the core, Mn increases again to 40 µM. Dissolved Ca concentrations show an overall downcore decrease from 9 to 5 mM (Fig. 3). Ammonium concentrations show a steady increase from 45 µM at 1 cm depth to 13.8 mM at 300 cm. The concentrations remain at this level for the rest of the core (Fig. 3). The pH averages are at 7.85 above and at 7.29 below the SMT, and DIC increases from 2.5 mM in the bottom water to 66 mM at 476 cm sediment depth (Fig. 3).
3.2 Solid-phase composition
Total Fe contents (Fetotal) of GC HE443/10-3 were shown earlier by Aromokeye et al. (2020) and are available in the PANGAEA database (Aromokeye et al., 2018a; with core depths instead of sediment depth). Sequentially extracted Fe data as well as Mn and Al contents are available in PANGAEA (Henkel et al., 2024b).
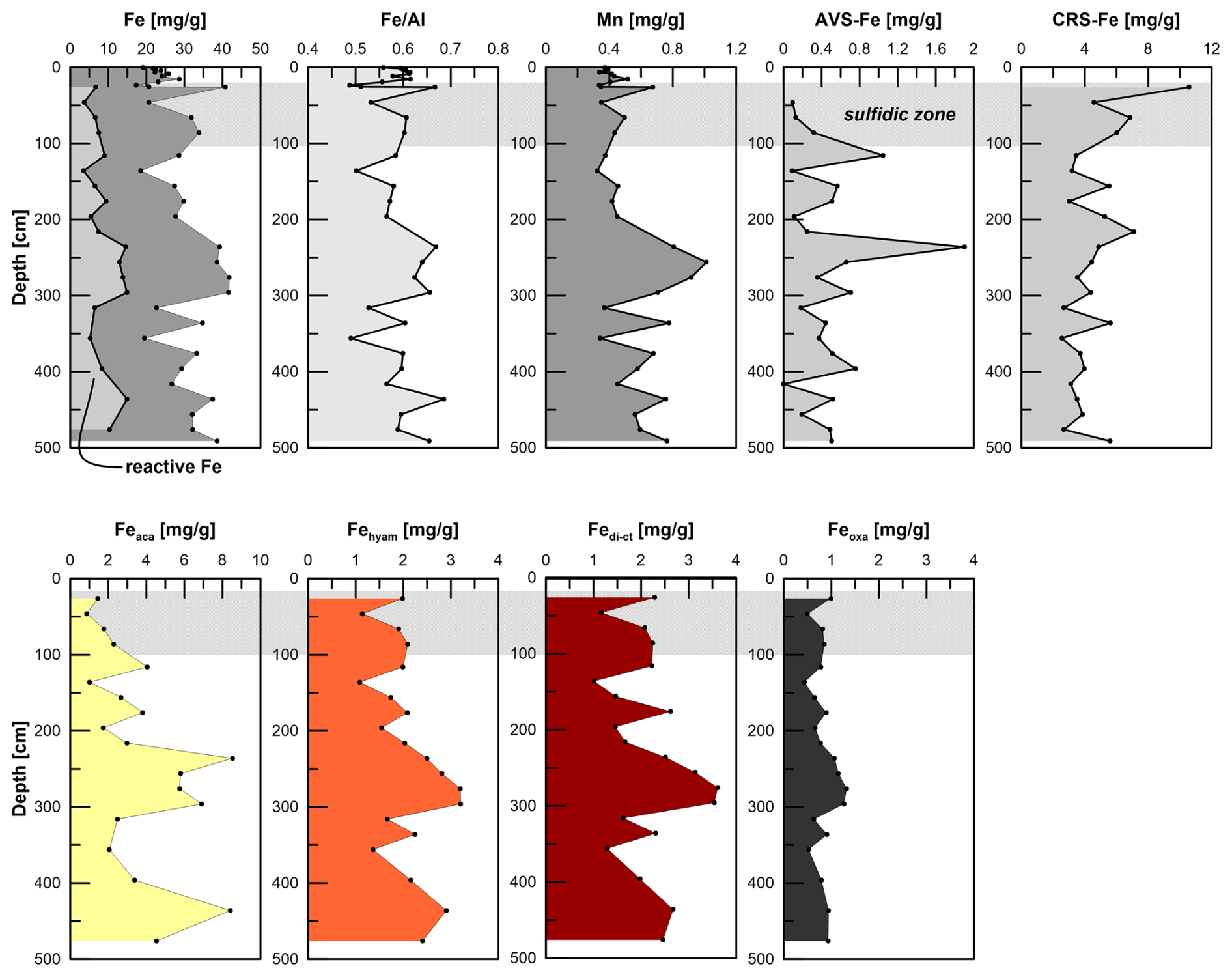
Figure 4Total and reactive Fe contents, , Mn, AVS–Fe, CRS–Fe, and sequentially extracted Fe pools after Poulton and Canfield (2005). Note that reactive Fe is the sum of Feaca (carbonates, surface-reduced Fe), Fehyam (amorphous easily reducible oxides), Fedith (goethite, hematite), and Feoxa (magnetite). Note that the sequential extraction is not mineral-specific but operationally defined.
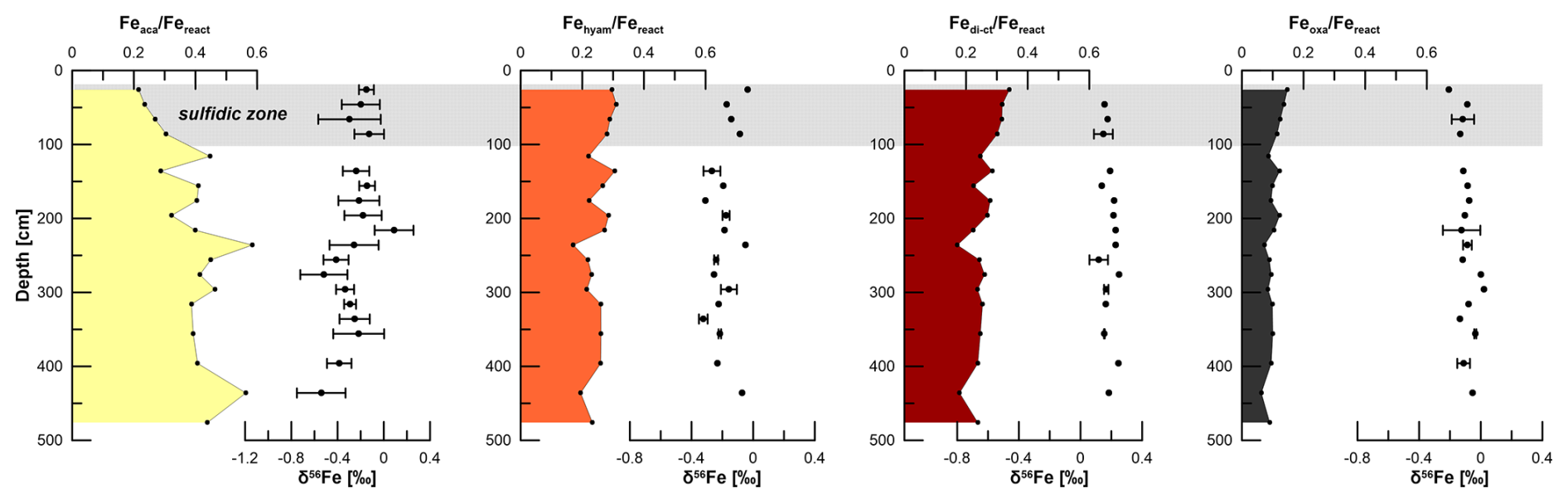
Figure 5Sequentially extracted Fe pools normalized to the sum of reactive Fe and the respective δ56Fe values. Uncertainty bars are 2 SD and given only for samples that were repeatedly analyzed. The grey bar indicates the sulfidic interval.
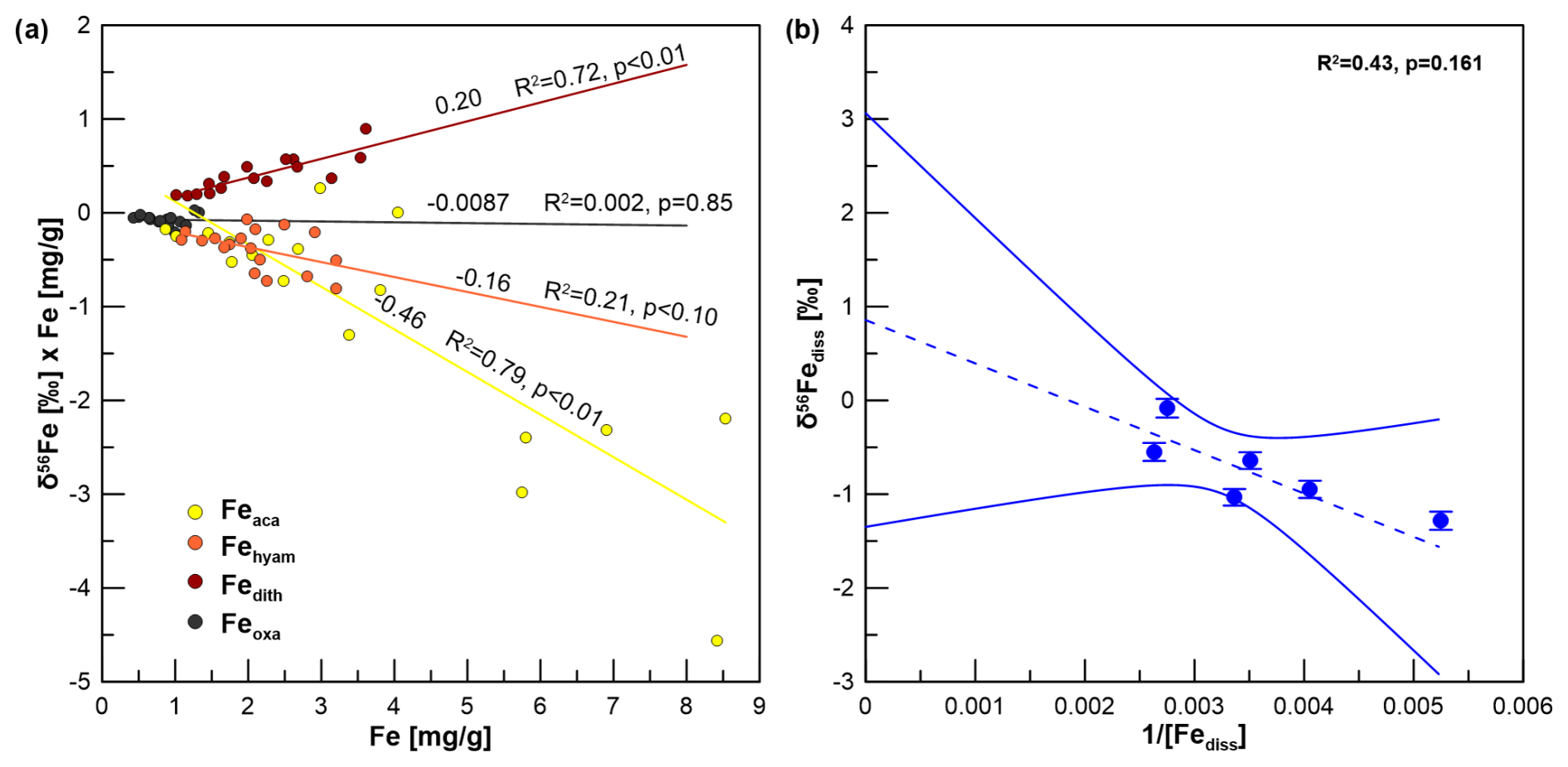
Figure 6(a) Miller–Tans plot for δ56Fe values of sequentially extracted reactive Fe pools. (b) Keeling plot for δ56Fe values of pore water with the 95 % confidence interval. We only used data from between 450 and 150 cm, where there is a rather linear δ56Fediss trend.
Fetotal varies between 17 and 42 mg g−1 (Fig. 4). The ratio (g g−1) at site HE443/10 is between 0.49 and 0.69 (average 0.59), with higher values corresponding to high Fetotal contents. According to the sequential extraction data, 16 %–30 % of Fetotal is associated with Fe-carbonates, FeS (which is not targeted here but dissolves in 1 M Na-acetate and was targeted in a separate extraction of Fe sulfides; see below), and Fe-oxides. There is no clear downcore decrease in Fetotal or the sequentially extracted Fe pools (Fig. 4). On the contrary, there are intervals of elevated Fe contents at 230–300 cm and below 400 cm, which are reflected by all extracted Fe phases. Only when plotted relative to reactive Fe (sum of Fe extracted by MgCl2, Na-acetate, hydroxylamine–HCl, Na-dithionite/Na-citrate, and ammonium oxalate/oxalic acid), the acetate-leached Fe pool (Fe-carbonates and surface-reduced Fe(II)) shows an overall increase with sediment depth, whereas the other extracted Fe fractions rather show a decrease (Fig. 5). Close to the sediment surface, the composition of reactive Fe is as follows: 20 % acetate-leached Fe (Feaca), 30 % hydroxylamine–HCl-leached Fe (Fehyam), 35 % dithionite-leached Fe (Fedi-ct), and 15 % oxalate-leached Fe (Feoxa). At 450 cm, the respective values are 50 % Feaca, 20 % Fehyam, 20 % Fedi-ct, and 10 % Feoxa. Only the Feaca pool shows a clear variation in the δ56Fe composition, where high Feaca contents correspond to low δ56Feaca signals down to −0.54 ‰ (Fig. 5). Data representation in a Miller–Tans plot (Miller and Tans, 2003; Fig. 6a), which allows assessing even small isotopic variations that depend on the size of the respective pool, underlines this relationship with R2 = 0.79 (p < 0.01). The hydroxylamine–HCl-leached pool shows overall negative δ56Fehyam values (−0.19 ± 0.17 ‰, 2 SD). As with the Feaca pool, the Miller–Tans analysis indicates a correlation between Fehyam content and the isotopic composition (Fig. 6a), with higher contents being related to more negative δ56Fehyam values. The relationship is, however, less clear (R2 = 0.21, p < 0.1), and the overall range of δ56Fehyam (−0.32 ‰ to −0.03 ‰) is considerably smaller compared to Feaca. δ56Fedi-ct values (Fig. 5) range between 0.12 ‰ and 0.25 ‰ (0.19 ± 0.08 ‰, 2 SD). The Miller–Tans plot indicates that there is a slight enrichment of 56Fe in samples that show high Fedi-ct contents (R2 = 0.72, p < 0.01) (Fig. 6a). Oxalate-leached Fe shows δ56Feoxa values of −0.09 ± 0.10 ‰ (2 SD) and neither a downcore trend (Fig. 5) nor a dependency of δ56Feoxa on Feoxa contents (Fig. 6a).
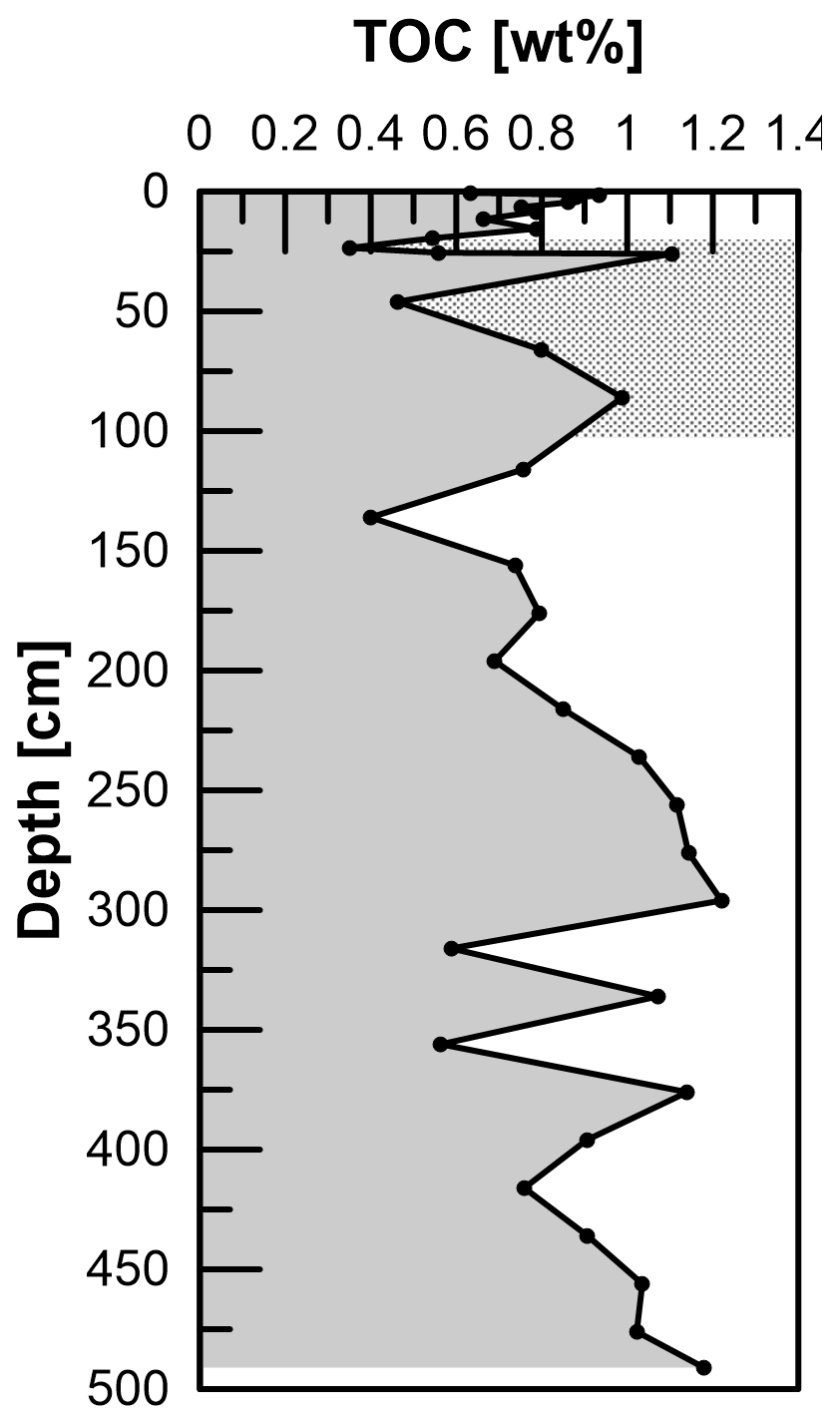
Total Mn contents in the solid phase range from 0.3 to 1 mg g−1, where the overall profile shape is very similar to the distribution of Fetotal: maximum values occur between 200 and 300 cm. Local maxima further downcore coincide with maxima in Fe at 335, 375, 435, and 490 cm. TOC values range between 0.4 wt % and 1.2 wt % (Fig. 7) and show a strong positive correlation with Fetotal (R2 = 0.76, p < 0.01). As for Fetotal, there is no decrease in TOC with depth but a zone of elevated values between 230 and 300 cm and a trend towards higher values (up to 1.2 wt %) towards the end of the core. Total inorganic carbon (TIC; data available under Henkel et al., 2024b) shows similar trends, but values are higher (1.3 wt % to 2.3 wt %) compared to TOC.
Iron sulfides are present as AVS and CRS. Both sulfide pools are not limited to the sulfidic zone (Fig. 4) but occur over the whole gravity core length. AVS peaks at the depth of the current sulfidization front at ∼ 100 cm depth (1 mg g−1 AVS–Fe). A second maximum of 2 mg g−1 AVS–Fe appears at 250 cm. CRS is highest at 26 cm depth (10 mg g−1 CRS-bound Fe), the uppermost sample that was analyzed. The contents decrease towards ∼ 120 cm (3 mg g−1 CRS-bound Fe) and remain at a level of ∼ 4 mg g−1 further downcore. AVS is affected by the extraction of Fe-carbonates with Na-acetate (Cornwell and Morse, 1987; Poulton and Canfield, 2005). Based on the separate extraction, the contribution of AVS to the Feaca pool was calculated to be between 6 % and 26 % (average 13 %). This is particularly important with respect to δ56Feaca data. Even though there is no correlation between the AVS contribution to Feaca (in %) and the respective δ56Feaca value (R2 = 0.03), δ56Feaca should be interpreted with caution as it represents a mixed signal of several (secondary) Fe pools that likely have different isotopic compositions: siderite, Fe monosulfides, and surface-reduced Fe(II) (Crosby et al., 2005, 2007; Henkel et al., 2016).
3.3 Model results (transport-reaction simulation)
We performed sensitivity tests by applying different fractionation factors α1 for DIR (Reaction R1, k1 as a function of OM degradation), different reaction rate constants k2 and a fractionation factor α2=1 for the sulfidization via the reaction of hydrogen sulfide with Fe-oxides (Reaction R2), and different reaction rate constants k3 and fractionation factors α3 for sulfide precipitation via reaction of Fediss with HS− (Reaction R3). The differences between the measured and calculated concentrations/values and at each depth i were calculated using the mean square error (MSE) as
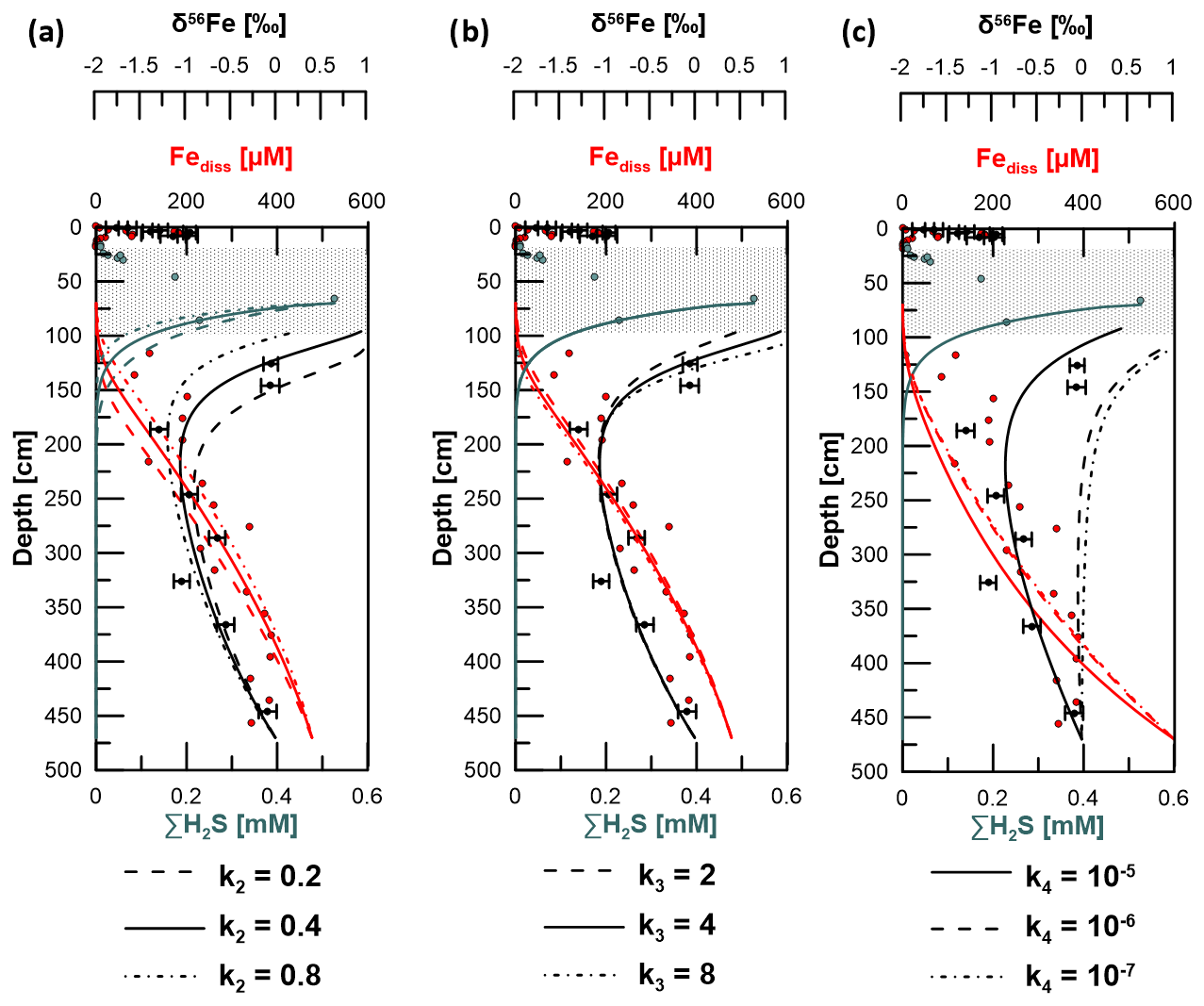
Figure 8Sensitivity test by application of different reaction rate constants (k) for (a) Reaction (R2) (reaction of free hydrogen sulfide with solid Fe-oxides), (b) Reaction (R3) (iron sulfide formation by reaction of Fe2+ with HS−), and (c) Reaction R4 (adsorption of Fe2+). Solid lines represent the best fit to the measured data and were thus used in the model to determine kinetic fractionation factors (see Table S2). Uncertainty bars are 2 SD.
The minimized sum of the MSE for Fe2+, HS−, and δ56Fediss was used to find the best-fitting parameters. The respective constants k2 applied for Reaction (R2) were 0.2, 0.4, and 0.8. The best fit based on the minimum MSE was achieved with k2=0.4 (Fig. 8). The reaction front of Fediss and HS− varies with the value k2. Although there is no isotopic Fe fractionation considered for Reaction (R2), the changed H2S profile leads to a different depth for Reaction (R3) and, thus, a different δ56Fediss profile. The constants k3 tested for Reaction (R3) (in combination with k2=0.4) were 2, 4, and 8. The H2S concentration profile shows a higher dependency on Reaction (R2) (or k2) compared to Reaction (R3) (or k3). The best data fit resulted from applying k3=4.
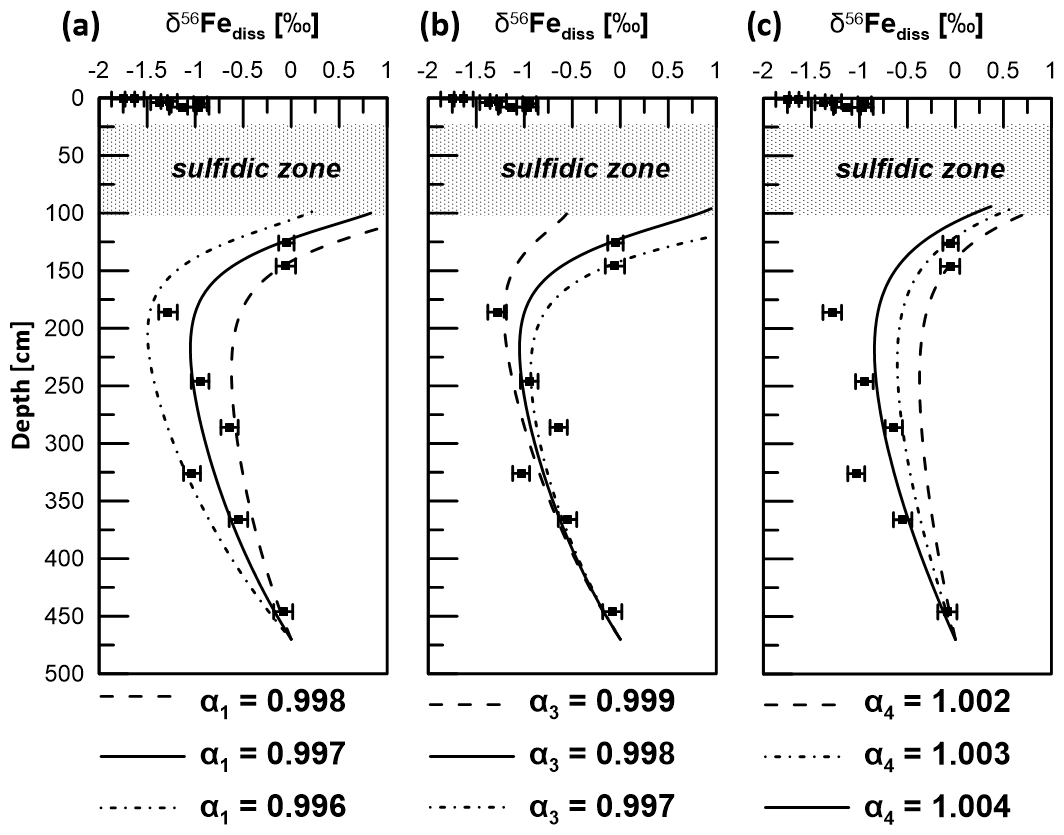
Figure 9δ56Fediss profiles derived by applying a transport-reaction model and different kinetic fractionation factors for (a) microbial iron reduction (α1), (b) pyrite formation via Fe2+ and HS− counter-diffusion (α3), and (c) adsorption of Fe2+ (α4). Solid lines represent the best fit to measured data (filled circles). Uncertainty bars are 2 SD.
Using k2=0.4 and k3=4 for Reactions (R2) and (R3), the best fit of the modeled δ56Fediss profiles with the respective measured data was achieved when setting the kinetic fractionation factor α1 for MIR (DIR) (Reaction R1) to 0.997 and α3 for sulfide precipitation via reaction of Fe2+ with HS− (Reaction R3) to 0.998 (Fig. 9). As Reaction (R1) is not confined to a narrow interval as is the case for Reaction (R3), the effect of the choice of α1 on the overall δ56Fediss profile is much stronger than the choice of α3. The value chosen for α3 is, of course, most relevant to the depth of ∼ 100 cm, where HS− formation occurs. Here, the choice of the fractionation factor results in differences in δ56Fediss of more than 2 ‰ (Fig. 9; modeled profile with α3=0.997 not shown completely due to the limitation of the x axis to 1 ‰).
4.1 Redox zones, reaction fronts, and sediment composition
The kink shape of the sulfate profile indicates some bioturbation and/or bioirrigation in the top 15 cm of the sediment (e.g., Henkel et al., 2011; Fischer et al., 2012), which is consistent with the general intense bioturbation at site HE443/10 evidenced by the radiographs (Henkel et al., 2024a). However, this does not result in a high O2 penetration depth as demonstrated by the presence of dissolved Mn (∼ 35 µM) at 1 cm and the maximum concentration at 3 cm (Fig. 3). Manganese oxides are considered to become microbially reduced as soon as the pore water is depleted of more favorable electron acceptors such as O2 and nitrate (e.g., Burdige, 1993). The Fediss profile (Fig. 2) is smooth and shows comparatively high concentrations of up to 180 µM at 5 cm depth. Therefore, we consider the direct effect of bioturbation and bioirrigation on pore-water geochemistry to be minor. The Fe liberation in this interval is mainly due to MIR, as has been described in detail in Henkel et al. (2016) for a site located ∼ 100 m away from site HE443/10.
The investigated sediments are rich in total and reactive Fe. The averages are 28 and 9 mg g−1 of sediment, respectively (Fig. 4). The ratio is ∼ 0.59, which is close to the average shale composition of 0.55 (Wedepohl, 1991). There is no downcore decrease in Fetotal, , or the reactive/extractable Fe pool as seen for example in Thamdrup et al. (1994) in sediments of the Bay of Aarhus (Denmark) and Severmann et al. (2006) in deposits of the Santa Barbara Basin. In the case of constant accumulation rates and a consistent composition of the accumulated material, a decrease in Fe or reactive Fe phases with depth is indicative of microbial Fe reduction, upward Fe2+ diffusion, and Fe-oxide precipitation at the redox boundary. The absence of such a decrease at site HE433/10 is likely due to intense reworking of the sediment. Bioturbation results in very effective mixing of solid phases in the top few centimeters.
Under the assumption that the ratio of the detrital material transported to the HMA area has been constant over time, intervals with high ratios and Fe contents (e.g., 230–300 cm) reflect enrichments of Fe due to diagenetic Fe mineral formation. In contrast, the intervals of lower and Fetotal contents indicate loss of Fe through early diagenetic Fe reduction and subsequent diffusion. However, Fe-oxide dissolution and secondary mineral formation might have been blurred in the record if they happened while the sediment was still in the zone affected by bioturbation. The release of Fediss into the pore water, in particular at ∼ 5 and 400 cm depth, and the presence of Fe monosulfides and pyrite in the whole sediment column generally reflect the fact that Fe phases at site HE443/10 undergo considerable early diagenetic transformation. Iron sulfides are indicative of the reaction of solid Fe(III) or Fe2+ with hydrogen sulfide, which is released during organoclastic or methane-mediated sulfate reduction (e.g., Poulton et al., 2004; Jørgensen and Kasten, 2006; Riedinger et al., 2017). The peak in H2S indicates that sulfate-mediated AOM takes place at ∼ 70 cm. There is a higher diffusive flux Jsed of HS− (ca. −13 ) compared to Fediss (0.60 ) towards the sulfidization front at 100 cm (Fig. 2). Consequently, there is not only precipitation as FeS, but also formation of pyrite from these monosulfides and Fe-oxides: the removal of HS− from pore water by sulfide formation or reoxidation exceeds the removal of Fediss.
The most reactive Fe-(oxyhydr)oxides with respect to H2S are hydrous ferric oxides, ferrihydrite, and lepidocrocite, followed by goethite, magnetite, and hematite (Findlay et al., 2020; Michaud et al., 2020; Poulton et al., 2004). The comparably high Fehyam contents at site HE443/10 indicate that the amount of H2S has never been high enough or the time the sediment was subjected to sulfidic conditions has never been long enough to lead to a complete transformation of the highly reactive Fe-oxide pool into iron monosulfides or pyrite. The high Fehyam contents are at least partly attributed to lepidocrocite, which has previously been detected by Mössbauer spectroscopy in methanic sediments of the HMA (Oni et al., 2015a). It needs to be noted that recent incubation studies showed that the term “reactive” Fe-oxides as we use it here based on chemical extraction is not necessarily identical to the fraction that is “biologically available” (Aromokeye et al., 2020; Wunder et al., 2024).
There is an enrichment in Fe sulfides (mostly CRS) within the current sulfidic zone and in particular at or close to the upper sulfurization front at 10 cm depth, where Fediss and H2S react to FeS, which is subsequently transformed into pyrite (Fig. 4). This enrichment shows that despite the strong reworking of the sediment, the sulfidic zone must have been fixed to this interval for some years, which is consistent with the low sedimentation rates of < 3 mm yr−1 during the past ∼ 700 years. The abovementioned Fetotal enrichment between 230 and 300 cm is not related to a CRS maximum, so it does not represent a paleo-SMT. It is rather attributed to the Feaca pool, potentially indicating a diagenetic formation of Fe-carbonates (siderite) or AVS. (At site HE443/10, siderite is oversaturated below 100 cm depth; see Fig. S1 in the Supplement.) The Fe enrichment is also, to a lesser extent, reflected by Fehyam and Fedi-ct, which indicates that this interval either has been “recharged” a lot with diagenetic Fe-oxides by oxidation of the upwardly diffusing Fe2+ when this interval was close to the surface or was buried more rapidly than sediment above and below so that Fe-oxides were not affected as much by pyritization. Since the interval does not show stronger signs of bioturbation than the rest of the core, which could favor a strong reoxidation of Fe2+ and FeS, and since sedimentation rates were high when this interval accumulated, we consider the faster burial through the sulfidic zone to be the more plausible explanation for the Fe enrichment observed between 230 and 300 cm.
The extraction with sodium acetate targets Fe associated with carbonates (Poulton and Canfield, 2005; Tessier et al., 1979) but is known to also partly dissolve, for example, monosulfides (AVS) (Cornwell and Morse, 1987; Poulton and Canfield, 2005) and surface-reduced Fe(II) (Crosby et al., 2005, 2007; Henkel et al., 2016). As we extracted AVS separately, we could attribute 6 % to 26 % of the Feaca pool in samples from site HE443/10 to AVS. Particularly high contributions of AVS to Feaca are found at 116 cm, just below the current depth of the lower sulfidization front; at 236 cm (depth of the abovementioned Fe enrichment); and at 396 cm. The presence of AVS below the sulfidic zone is consistent with the findings of Riedinger et al. (2017) that show that metastable authigenic iron monosulfides can survive burial into deeper sediments in highly dynamic depositional systems, where high sedimentation rates and high Fe-oxide contents limit the exposure to free HS−, which is restricted to a narrow zone around the SMT.
Diagenetic processes and reaction fronts in marine sediments are ultimately determined by the quality and amount of accumulated TOC (e.g., Rullkötter 2006). In order to assess whether reaction fronts might have shifted upwards or downwards in the past, in particular as a consequence of the abovementioned decrease in sedimentation rates and a potential change in organic matter accumulation, we determined TOC contents. The preserved TOC contents at site HE443/10 are all below 1.2 wt %. Considering some uncertainty as our study site is 5 km southwest of the core dated by Hebbeln et al. (2003), the shift in sedimentation rates should relate to a depth between 1 and 2 m at HE433/10. Even though TOC values scatter between 0.4 wt % and 1.2 wt %, there is no clear shift that would hint at this drastic change in depositional conditions. We conclude that the overall composition of material that was deposited in the HMA before and after ∼ 700 BP was similar and that the change was largely limited to the amount of material that was supplied. This is in line with previous observations by Oni et al. (2015b), who suggested similar sources of sediments and organic matter at and below the depth of the SMT based on low variation in δ13C of TOC.
4.2 Iron isotope fractionation
As in other marine environments, where in situ Fe reduction in shallow sediments has been observed (e.g., Henkel et al., 2016, 2018; Homoky et al., 2009; Severmann et al., 2006), there is, above the sulfidic zone, an overall downcore trend of δ56Fediss towards heavier values (−1.75 ‰ at 1 cm vs. −1 ‰ at 8 cm). This trend is related to (1) preferential removal of light Fe isotopes from the reducible ferric Fe pool during burial and ongoing MIR as well as to (2) preferential removal of light Fe isotopes during interactions with hydrogen sulfide at the sulfidization front (Severmann et al., 2006). The availability of 54Fe(III) is highest close to the oxic–anoxic boundary, which is reflected by the most negative δ56Fediss in the pore water. The processes above the sulfidic zone of HMA sediments were described earlier by Henkel et al. (2016). The easily reducible Fe(III) pool (Fehyam) contains Fe-oxides that formed from isotopically light Fediss and is therefore also isotopically light compared to less reactive Fe-oxide pools. This has already been shown for shallow HMA sediments (Henkel et al., 2016) but can also be observed for the deeper sediments investigated here (δ56Fehyam −0.19 ± 0.16 ‰ (2 SD) compared to δ56Fedi-ct 0.19 ± 0.08 ‰ and δ56Feoxa −0.09 ± 0.10 ‰).
Dissolved Fe concentrations measured right below the sulfidic zone, at ∼ 130 cm, are ∼ 100 µM. Respective δ56Fediss values are ∼ 0 ‰ (compared to −1.28 ‰ at ∼ 190 cm, from where Fe is diffusing upwards). At first glance, this is in accordance with observations by Severmann et al. (2006) in sediments from Monterey Bay and Santa Barbara Basin and in sediments from Lake Kinneret (Sivan et al., 2011), where the formation of amorphous Fe sulfides drives δ56Fediss towards positive values by preferential removal of 54Fe from pore water. Experimental studies show that the δ56Fe composition of FeS can be highly variable and depends on proportions of isotope exchange between particle and Fediss during aging of iron monosulfides (Guilbaud et al., 2010). Nevertheless, there is general agreement that kinetic isotope fractionation, which dominates in natural sediments, leads to an isotopically light composition of amorphous Fe monosulfides compared to Fediss with = −0.85 ± 0.30 ‰ ( = 0.999) (Butler et al., 2005; Roy et al., 2012). The 54Fe is further preferentially incorporated into pyrite (with FeS as precursor), as was shown by Guilbaud et al. (2011) for abiotic pyrite formation. Here, is −1.70 ‰ to −3.0 ‰ ( = 0.998 to 0.997), so the combined fractionation factor (that can be compared to our α3=0.998, Fig. 9b) is 0.996 to 0.997. It needs to be considered that Fe sulfides age and exchange Fe isotopes with their surroundings (equilibrium fractionation). This is a process which takes place continuously in marine sediments. It might not be dominant, especially not at reaction fronts, but it causes a continuous equilibration of the Fe isotopic composition of different pools, also below the sulfidic interval.
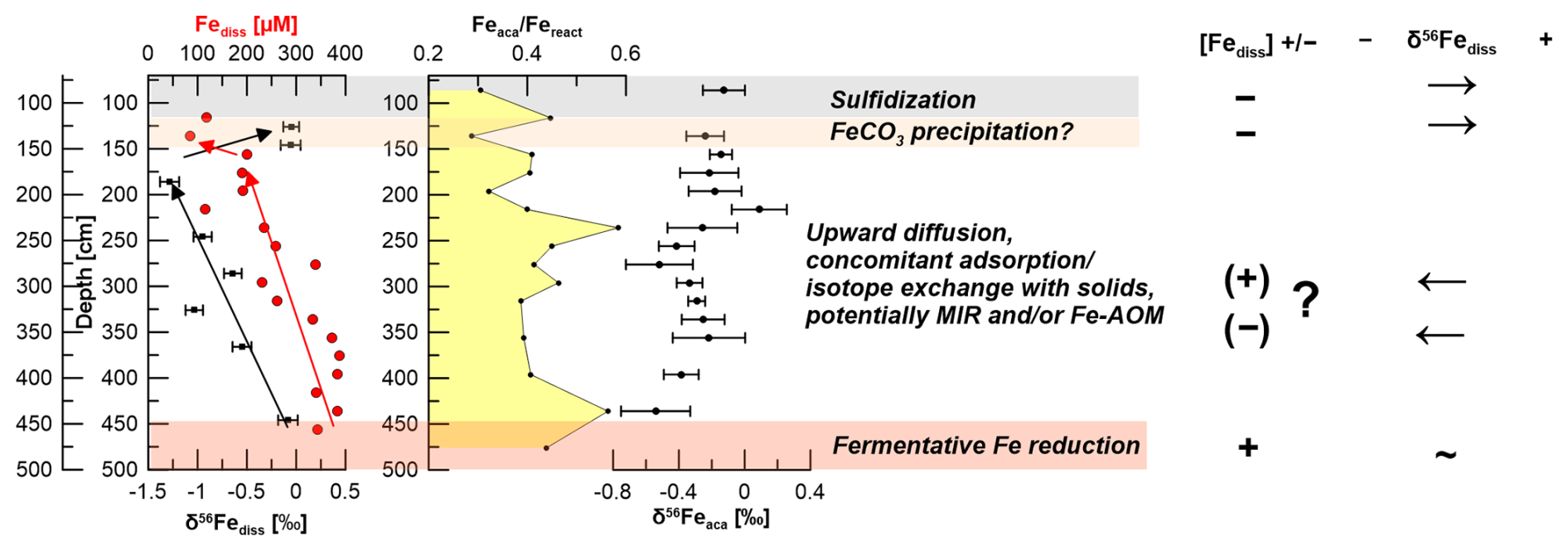
Figure 10Interpretation of pore-water and stable Fe isotopic data for site HE443/10: the deep Fe release does not coincide with significant Fe fractionation and might be explained by fermentative processes. The red arrows show the overall trend of Fediss. The black arrows show the respective trends of δ56Fediss values. The upward trend towards lighter isotopic values (between 450 and ∼ 175 cm) might be explained by either low rates of microbial iron reduction (MIR), preferential removal of 56Fe by adsorption, or both. Additionally, Aromokeye et al. (2020) showed low rates of Fe-AOM in incubations of methanic HMA sediments. This process might also affect δ56Fediss. The shift towards positive δ56Fediss values at ∼ 150 cm likely indicates siderite precipitation.
At a second glance, however, it becomes apparent that there is a mismatch between the modeled Fe2+ and HS− profile for this process at site HE443/10 and the respective measured data (Fig. 8b). From its source (AOM at ∼ 70 cm depth) HS− diffuses downwards and is used up already at a depth of ∼ 120 cm, where Fediss is already at 118 µM. Based on the model output, HS− would diffuse down to a depth of almost 150 cm and Fediss concentrations at ∼ 130 cm would be close to zero. Furthermore, although we want to be cautious not to over-interpret a single data point, an Fediss sink is indicated by our measured pore-water profile at ∼ 135 cm (loss of ∼ 50 % of the upwardly diffusing Fe). We conclude that siderite precipitation might occur at this specific interval. As for the precipitation of FeS, siderite formation would preferentially transfer 54Fe into the solid phase ( = −0.48 ± 0.22 ‰; Wiesli et al., 2004). Unfortunately, the Feaca contents vary overall too much to resolve where there is or has been a particular interval affected by siderite formation, and δ56Feaca in this potentially affected interval is similar to the δ56Feaca directly above and below (Fig. 10). DIC concentrations are too high (> 50 mM) to reflect a sink on the order of less than 2 as calculated from the loss of Fediss at 135 cm.
A Keeling plot (Fig. 6b) was used to determine the pore-water Fe isotope endmember for the observed deep Fe release. Here, we only used data from below those depths at which δ56Fediss is mainly controlled by the reaction with H2S, i.e., between 450 and 150 cm, where there is a rather linear δ56Fediss trend (see Fig. S2 in the Supplement for reaction rates of Reactions R1–R3). Although there is a linear trend between and δ56Fediss (R2 = 0.43), the correlation is not statistically significant (p value 0.161), which is partly due to the low number of data points. The 95 % confidence interval covers a wide range between −1.4 ‰ and +3.0 ‰ for the inferred Fe source (the intercept with the y axis); it is not possible to determine the endmember without a large error. However, the Fe liberation at depth is most likely not causing a preferential release of 54Fe. The lowermost δ56Fediss value is −0.08 ± 0.10 ‰ and has thus an isotopic composition which is similar (within uncertainty) to the composition of Fehyam and Feoxa and is only slightly lighter than Fedith (Fig. 5).
The δ56Fediss value at ∼ 190 cm is −1.28 ± 0.10 ‰ (2 SD), so while diffusing upwards, the Fediss either (1) loses heavy isotopes or (2) is affected by an additional process providing light isotopes. Removal of 56Fe from pore water happens by preferential adsorption onto (Fe-oxide) particles, as has recently been shown by Köster et al. (2023). Over time, this Rayleigh distillation process progressively lowers δ56Fediss values. Köster et al. (2023) used the fractionation factors = 0.87 ‰ and 1.24 ‰ for adsorption of Fe(II) onto goethite surfaces (Beard et al., 2010; Crosby et al., 2007) to calculate the proportion of Fediss that would need to be “removed” (adsorbed) in order to obtain the extremely negative δ56Fediss values that they measured in deep, lithified sediments from the Nankai Trough. Using the same approach here with δ56Fediss = 0 ‰ for the deep Fe source, between 65 % and 75 % of the Fediss pool would need to be adsorbed to achieve a value of −1.28 ‰ at 190 cm. The Fediss concentration at ∼ 190 cm is about 50 % of [Fediss] at 400 cm (Fig. 2). It is likely that adsorption (and also electron and atom exchange with different Fe minerals) takes place, which means that the concentration profile of Fediss in the methanic zone is not solely controlled by Fediss release at depth and Fe–S reactions as a sink. Therefore, by implementing a reaction for adsorption based on Wang and Van Capellen (1996; see Table S1) and the rate expression with , 10−6, or 10−7 in our model, we tested if adsorption of Fe2+ plays a significant role (apart from being already included into the DIR fractionation factor α1 as described in Sect. 2.5). k4 includes the number of unoccupied surface sites, which is unknown. The test results do not produce a good fit between the modeled and measured Fediss profiles (Fig. 8c). Including α4=1.002, 1.003, or 1.004 for the respective preferential removal of the heavy isotope (in combination with ) also did not lead to a good reproduction of the observed distinct shift in δ56Fediss to −1.28 ‰ at ∼ 190 cm (Fig. 9). Furthermore, the adsorbed (heavy) Fe should be transferred to the Feaca pool, but δ56Feaca instead shows a trend towards more negative δ56Feaca values with depth (Fig. 10). We conclude that adsorption in the methanic sediments at site H433/10 is not the process dominating Fe removal from pore water and the trend towards lighter δ56Fediss values between 450 and ∼ 190 cm.
Our PHREEQC calculations also indicate vivianite saturation in the methanic sediments. Vivianite is a ferrous iron phosphate mineral, Fe3(PO4)2 ⋅ 8 H2O, which is known to be stable under anoxic sedimentary conditions, but its identification and quantification are difficult. Rothe et al. (2014) found that supersaturated pore water alone does not reliably predict vivianite formation. Furthermore, there are no data available yet concerning the fractionation of iron during vivianite formation. Vuillemin et al. (2020) determined negative δ56Fe values in vivianite crystals in more than 20 m deep sediments from Lake Towuti, Indonesia, but the dataset does not include pore-water data. We conclude that we do not have enough data to assess the role of vivianite precipitation at our study site. However, vivianite formation would act as a phosphorous sink, and the PO4 profile indicates a sink at ∼ 300 cm. A slight shift in δ56Fediss values towards negative values is recorded slightly below at 325 cm (Fig. 10). Therefore, one could speculate that vivianite precipitation happens and preferentially incorporates 54Fe.
For the model, we tested MIR as the factor controlling the trend of δ56Fediss values towards −1.28 ‰ at between 450 and ∼ 190 cm depth. We did so because MIR was stimulated in incubations of methanic sediments from this site when easily reducible Fe-oxides (lepidocrocite) and benzoate were provided (Aromokeye et al., 2021). Therefore, Fe reducing bacteria are present in these sediments, and Oni et al. (2015a) have also shown the presence of lepidocrocite. We also see a concurrent net Mn release in methanic sediments between 150 and 200 cm but only at low levels, potentially because Mn solubility in non-sulfidic sediments is generally controlled by rhodochrosite or other mixed -carbonates (e.g., Gingele and Kasten, 1994). The net Mn release hints at microbial metal reduction (overlap of MnO2 reduction and MIR). In natural methanic sediments, organoclastic DIR with fermentation intermediates such as acetate is generally assumed to not play an important role because the related bacteria need acetate or other intermediates of fermentation products, which are typically not available at these depths. However, a recent study at a comparable site in the Bay of Aarhus (Kattegat) showed higher acetate concentrations below the SMT than above (Glombitza et al., 2019). Furthermore, through microbiological enrichment experiments, Aromokeye et al. (2021) demonstrated the potential of Fe liberation in the deeper sediments of the HMA being related to the activity of fermenting bacteria, and those are known to produce acetate (Lovley and Phillips, 1986). The OM in methanic sediments was previously characterized as recalcitrant O-rich aromatic and highly unsaturated compounds of terrestrial origin (Oni et al., 2015b), but the authors' methods could not resolve the distribution of low-molecular-weight compounds such as short-chain fatty acids (including acetate). Microbes specialized in recalcitrant aromatic OM degradation often require initial fermentation of the OM to fermentation intermediates (e.g., volatile fatty acids or reducing equivalents, i.e., H2 and acetate) that can be accessed by dissimilatory iron-reducing organisms. In the methanic zone, fermentation intermediates such as acetate and H2 are likely electron donors for methanogenesis, whereas in surface sediments, organic fermentation products are often the electron donors for anaerobically respiring microorganisms with available electron acceptors (sulfate, iron oxides) (e.g., Beulig et al., 2018; Jørgensen, 2006; Whiticar, 1999; Yin et al., 2024). In our model, the applied MIR rate that produces a good fit to the measured δ56Fediss data is very low (0.00011 mM yr−1, Fig. S2) and does not explain Fediss concentrations of almost 400 µM at depth. When MIR takes place, the reactive ferric pool typically decreases with depth and becomes enriched in 56Fe. The rate is, however, too low to reflect this in the solid-phase data. The reactive Fe pool does not decrease with depth, and downcore isotopic trends are also absent for Fehyam, Fedith, and Feoxa (Fig. 4). We used a simple calculation to test how strongly the isotopic composition of the ferric Fe pool (here Fehyam) would change just by MIR at the rate applied in our model. The δ56Fehyam at depth L can be calculated according to the mass balance equation:
where and are the weight percent of Fehyam at the sediment surface and depth L (m), respectively, with its Fe isotope value and . ΔFe is the amount of Fe that was lost due to Fediss release by MIR; i.e., ΔFe = With a sedimentation rate of ω = 0.0016 m yr−1 and an MIR rate of R1 = 0.00011 mM yr−1, ΔFe is calculated as follows:
where MFe is the molecular weight of iron. According to this, the Fehyam pool would lose only 0.014 wt % (0.14 mg g−1) between the sediment surface and 5 m depth. The isotopic difference in Fehyam between the surface and 5 m depth would be 0.1 ‰, which is in the range of our analytical uncertainty (2 SD).
We note that Aromokeye et al. (2020) also demonstrated that Fe-AOM occurs at low rates, in particular right below the sulfidic zone. This process that releases 8 mol of Fediss for each mole of CH4 might potentially also play a role. But as there are no literature data on the respective Fe fractionation, our study cannot resolve whether it is solely MIR or MIR and Fe-AOM occurring at low rates (Fig. 10).
We observe that high Fedith contents are related to slightly higher δ56Fedith values (Fig. 6a), a situation that is counterintuitive assuming that 54Fe would be preferentially lost when the Fedith pool is reduced by microbes, so lower contents should coincide with a more positive δ56Fedith value. The Miller–Tans plot should therefore not be interpreted as reflecting downcore trends: Fedith (as well as Feaca, Fehyam, and Feoxa) maxima are recorded within the methanic interval (Fig. 4). The plot demonstrates that the isotopic differences between sequentially extracted Fe pools are largest (but still small) where Fe contents are highest – a circumstance that is also unexpected since the isotopic fractionation in the (remaining) substrate should be expressed more strongly if the ferric Fe pool is small. This is possibly an effect of non-steady-state conditions in the past. Overall, the Feoxa pool is not affected by Fe isotope fractionation, but all other extracted Fe(III) pools are. The pools that are known to contain sorbed Fe(II) and typical secondary minerals (Feaca and Fehyam) form from (isotopically light) Fediss and are therefore characterized by overall negative δ56Fe values and, as expected, lighter composition at higher contents. Only the δ56Fedith pool shows a slight shift towards positive values at higher contents, which we cannot entirely resolve here. Considering that the iron released at depth has an isotopic composition close to 0 ‰, adsorbed iron deriving from upward diffusion would potentially have a positive δ56Fe signature. If part of the adsorbed (heavy) iron is then exchanged with the reactive Fe-oxide surface (Crosby et al., 2007) and might subsequently even migrate deeper into the iron oxide crystal (Larese-Casanova et al., 2023), it could cause an alteration of Fe-oxide isotope signatures towards positive values without reducing the mineral. It might also be speculated that adsorption and the related electron and atom exchange are more prevalent at depths that have a high Fe-oxide (Fedith) content, but this interpretation remains very speculative, in particular because our model does not indicate adsorption to be a dominant Fe sink.
4.3 Deep iron release
Deep iron reduction can occur purely abiotically via oxidation of reduced sulfur compounds (e.g., Holmkvist et al., 2011; Riedinger et al., 2010). However, Fe reduction due to this so-called cryptic S cycling fails as an explanation for the buildup of Fediss far below the sulfidic zone, as has also been concluded by Riedinger et al. (2014) for Fe-rich continental-margin sediments off Argentina, by Egger et al. (2014) for Bothnian Sea sediments, and by Oni et al. (2015a) for the methanic sediments of the HMA.
Respiratory methanogenic iron reduction (e.g., Sivan et al., 2016; Eliani-Russak et al., 2023; Gupta et al., 2024, and references therein) might be a possible explanation for deep Fe release in methanic sediments. However, as the process is respiratory, we assume it would lead to Fe isotope fractionation similar to MIR in shallow sediments. Furthermore, in order to perform respiratory Fe(III) reduction, methanogens would need to oxidize CH4 or an organic substrate, e.g., acetate or methyl compounds. It seems unlikely that methane oxidation would support growth coupled to Fe(III) reduction (see Chadwick et al., 2024). Gupta et al. (2024) stated for example that “even though we and others have shown that methanogens like M. acetivorans are metabolically active and can conserve energy by iron respiration …, it is still not known whether methanogens can couple iron reduction to growth in addition to energy conservation.” The correlation of Fediss concentrations with JS1 bacteria, methanogens, and Methanohalobium/ANME-3-related archaea at our study site suggested that the deep Fe reduction is coupled to the activity of these microbes (Oni et al., 2015a). An overlap of hydrogenotrophic CH4 production and Fe-AOM has also been proposed based on the isotopic composition of CH4 in sediments from the Baltic Sea (Egger et al., 2014, 2017). Several incubation experiments have demonstrated that the addition of reducible Fe-oxides can stimulate Fe-AOM in natural sediments characterized by low/absent sulfate concentrations (Aromokeye et al., 2020; Beal et al., 2009; Egger et al., 2014; Segarra et al., 2013; Sivan et al., 2011). Aromokeye et al. (2020, 2021), however, found that in incubations of HMA sediment, Fe release occurred not entirely through Fe-AOM but was largely unrelated to methane oxidation and seemed to be instead linked to the fermentation of complex organic matter – a process that can be stimulated by crystalline Fe-oxides because (1) fermenters reduce Fe(III) as an outlet for electrons primarily to overcome the thermodynamic barriers caused by high concentrations of newly produced fermentation intermediates, thus enabling continued OM degradation (fermentative iron reduction; Hopkins et al., 1995; Lovley, 1991), and (2) the conductive character of the Fe-oxides facilitates interspecies electron transfer from fermenting bacteria towards methanogens (Kato et al., 2012; Lovley and Holmes, 2022).
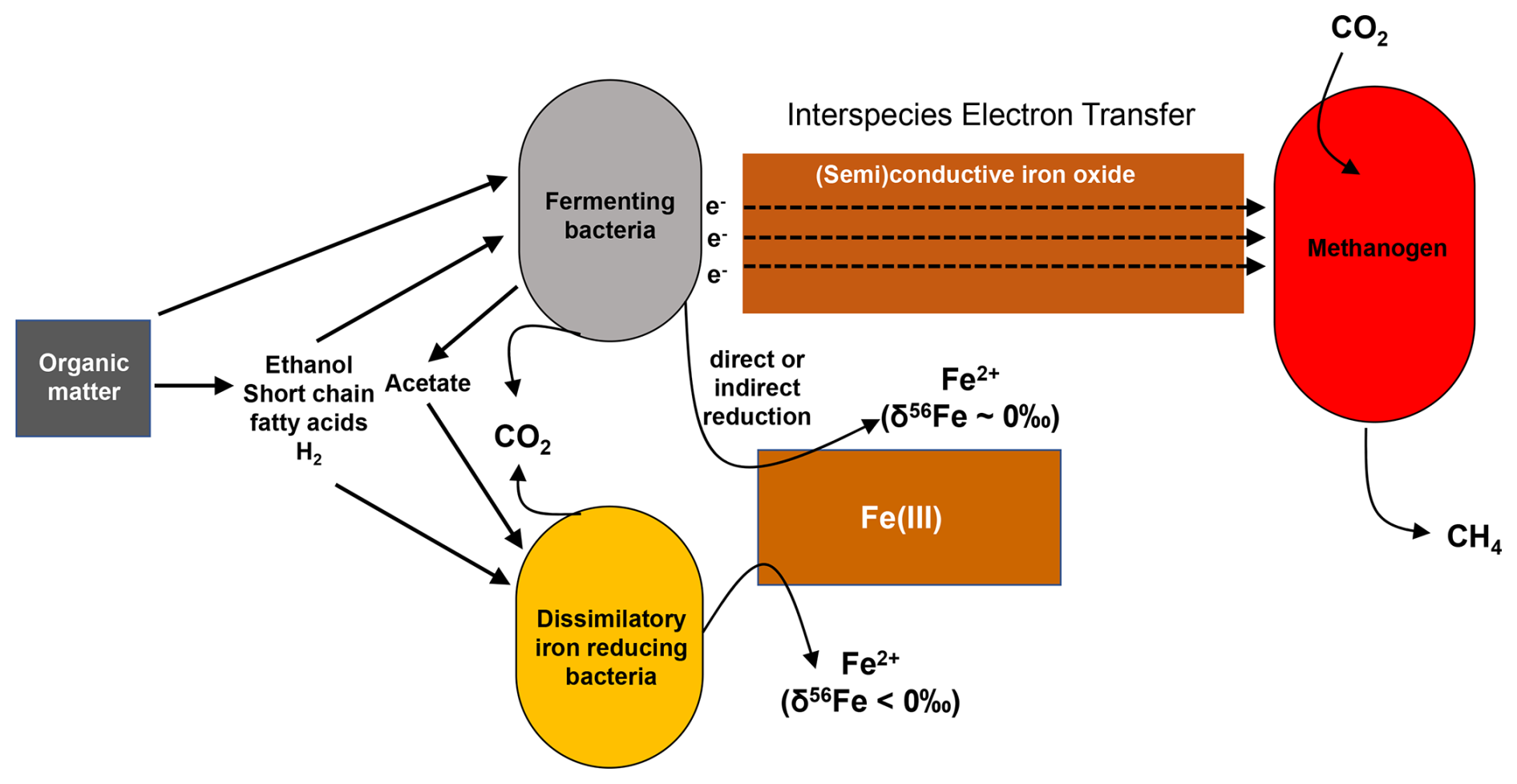
Figure 11Schematic representation of how deep iron reduction in methanic sediments of the HMA could be controlled. The relative contributions of microbial iron reduction (here DIR) and fermentative iron reduction likely depend on the availability of Fe-oxides, the composition of the organic matter, and the abundance of methanogens as “partner organisms” to which fermenters transfer electrons.
Building on all these previous studies, our iron isotopic data further hint at a deep Fe release that is not linked to DIR or another process in which microbes would preferentially use isotopically light Fe-oxides. Fermenting bacteria typically require a syntrophic partner such as an H2-utilizing bacterium (Hopkins et al., 1995) or a methanogen (e.g., Kato et al., 2012). The syntrophic partner consumes fermentation intermediates as a primary pathway for electron release and for thermodynamic feasibility of OM degradation. In the absence of a syntrophic partner, fermenting bacteria have been shown to be capable of electron transfer to iron oxides to further promote OM degradation. The iron oxides may be reduced fortuitously in the process as a final sink for the electrons or serve as a conduit, further transferring the electrons to an available syntroph, e.g., a methanogen (Aromokeye et al., 2021). The fermenting bacteria that transfer electrons to crystalline Fe-oxides do not directly profit from Fe(III) reduction beyond the removal of thermodynamic limitations brought about by accumulation of fermentation intermediates. In other words, the fermenters use the conductive Fe-oxides to transfer electrons and to be able to continue with the fermentation of particularly aromatic OM. The transfer of electrons via conductive Fe-oxides speeds up the degradation of aromatic compounds and is metabolically and mechanistically beneficial to both partner microbes (e.g., Jiang et al., 2013; Kato et al., 2012; Cruz Viggi, 2014; Zhuang et al., 2015). Meanwhile some doubt is building up that those electrons are really conducted in an electronic fashion without reduction and reoxidation of iron occurring. This is summarized in a review article by Xu et al. (2019). Figure 11 summarizes how fermenting bacteria, MIR-performing bacteria, and methanogens are known to interact and how the deep iron release as observed in the sediments of the HMA could be explained. It is known that in subseafloor sediments, there is a cooperative exchange of electrons and hydrogen in microbial communities and that this is also happening during fermentation (Shah et al., 2013). But to the best of our knowledge, there are no studies available that show how fermentative iron reduction takes place mechanistically, i.e., directly or indirectly by the fermenting bacteria or during the interspecies electron transfer. In addition, there are no experimental studies on how fermentative iron reduction fractionates iron isotopes. This is a gap in knowledge that should be addressed by future studies. In any case, the reason for the supposed fermentative Fe reduction happening at depth and not, for example, directly below the sulfidic zone might be selective OM degradation with more aromatic or unsaturated compounds in the deep sediment (Gibson and Harwood, 2012; Oni et al., 2015b).
4.4 Applicability of iron isotopes to trace iron processes in marine sediments
We demonstrate that the application of iron isotopes in marine sediments provides information that helps in identifying or verifying specific Fe reaction pathways. However, the main difficulty in using iron isotopes in natural systems is that, usually, various processes of Fe liberation and incorporation into solid phases are at play simultaneously. In the deep HMA sediments which contain lepidocrocite (Oni et al., 2015a) as well as crystalline Fe-oxides, different pathways of microbial organic matter oxidation with the involvement of Fe phases are likely to happen simultaneously – namely microbial iron reduction and fermentative processes with electron shuttling. These processes are therefore hard to resolve by iron isotope data alone. Generally, when multiple iron reactions are taking place, the resulting Fe isotope signals in dissolved and solid pools might not reflect or resolve all specific fractionation processes. Furthermore, over time, equilibrium isotope fractionation overprints kinetic isotope fractionation, so the isotopic composition of two pools that are susceptible to atom exchange will change until heavy isotopes are enriched in the pool with “stiffer” bonds (e.g., Wiederhold, 2015). We also note potential challenges when working with sequentially extracted Fe pools. As has been shown previously, these pools are not mineral-specific (e.g., Henkel et al., 2016). If the overall content of a pool is large compared to the amount of Fe in that pool that was affected by diagenesis, then the respective isotopic differences (e.g., downcore) might still be in the range of the analytical uncertainty. Therefore, depending on the setting, resolving differently reactive Fe phases and analyzing the respective δ56Fe signals might not be specific enough to clearly deduce which processes took place. This study and the comparison to the study of Köster et al. (2023) demonstrate that, unsurprisingly, processes dominating the shape of δ56Fe profiles and records differ depending on depth for two reasons: (1) the microbial community changes in composition and quantity due to depth-dependent availabilities of organic matter and electron acceptors and (2) equilibrium fractionation and processes like adsorption become increasingly important with the age of the studied sediment. In any case, the application of Fe isotopes in marine sediments requires a large set of complementary geochemical and microbiological data to achieve a robust interpretation.
Here we applied stable iron isotope analyses to pore water and sequentially extracted, differently reactive iron phases and transport-reaction modeling to identify the process responsible for the observed deep iron release in methanic sediments of the Helgoland mud area. The comparison between the isotopic composition of dissolved Fe and the ferric solid substrates reveals that the deep Fe release does not lead to a preferential liberation of 54Fe as occurs during DIR in shallow sediments. In combination with previous microbial studies, this isotopic study implies that iron reduction occurs during fermentative iron reduction when electrons are transferred from fermenters to iron oxides. In contrast to DIR, the “choice” of iron isotopes during the reduction seems to be rather coincidental. However, studies on the mechanistic details of fermentative iron reduction (including Fe isotope analyses) are needed to prove our interpretation.
This study provides a concept for how to deal with the complexity of geochemical and in particular Fe isotope data from pore water and sediments in order to test whether specific Fe redox reactions are or are not at play. We conclude that in combination with microbial experiments and geochemical and transport-reaction modeling, basic additional knowledge about Fe reactions can be gained by applying Fe isotope geochemistry. However, data interpretation is still far from being straightforward. This study also demonstrates that robust data interpretation relies on a combination of methods and the involvement of different areas of expertise.
Data are available in PANGAEA (https://doi.org/10.1594/PANGAEA.893760, Aromokeye et al., 2018a; https://doi.org/10.1594/PANGAEA.893766, Aromokeye et al., 2018b) linked to the publications by Aromokeye et al. (2020) and Henkel et al. (2024a, b) (DOIs: https://doi.org/10.1594/PANGAEA.971531, https://doi.org/10.1594/PANGAEA.971490).
The supplement related to this article is available online at https://doi.org/10.5194/bg-22-1673-2025-supplement.
SH and SK designed the study. MS, AM, and SAK provided expertise, access to MC ICP-MS instruments, and technical support. BL performed the modeling. SH compiled the geochemical data and wrote the manuscript with contributions from all co-authors. All co-authors were involved in data discussion.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We thank Ingrid Stimac (AWI), Ingrid Dohrmann (AWI), Jochen Scheld (University of Cologne), and Johann Hollop (at that time a student at the University of Bremen) for their support during sample processing. We particularly thank Clara Sena, who provided the thermodynamic database used in the PHREEQC calculations as well as the sample input files. Gerhard Kuhn (AWI) is thanked for providing TOC and TC data. We thank the two anonymous reviewers for their helpful comments and suggestions.
This research has been supported by the Deutsche Forschungsgemeinschaft (grant no. 390741603) and the Bundesministerium für Bildung und Forschung (grant no. 03F0874A).
The article processing charges for this open-access publication were covered by the Alfred-Wegener-Institut Helmholtz-Zentrum für Polar- und Meeresforschung.
This paper was edited by Chiara Borrelli and reviewed by two anonymous referees.
Arndt, S., Jørgensen, B. B., LaRowe, D. E., Middelburg, J. J., Pancost, R. D., and Regnier, P.: Quantifying the degradation of organic matter in marine sediments: A review and synthesis, Earth Sci. Rev., 123, 53–86, https://doi.org/10.1016/j.earscirev.2013.02.008, 2013.
Aromokeye, D. A., Kulkarni, A., Elvert, M., Wegener, G., Henkel, S., Coffinet, S., Eickhorst, T., Oni, O. E., Richter-Heitmann, T., Taubner, H., Wunder, L. C., Yin, X., Zhu, Q.-Z., Hinrichs, K.-U., Kasten, S., and Friedrich, M.: Solid phase geochemistry of sediment core HE443/10-3, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.893760, 2018a.
Aromokeye, D. A., Kulkarni, A., Elvert, M., Wegener, G., Henkel, S., Coffinet, S., Eickhorst, T., Oni, O. E., Richter-Heitmann, T., Taubner, H., Wunder, L. C., Yin, X., Zhu, Q.-Z., Hinrichs, K.-U., Kasten, S., and Friedrich, M.: Pore water geochemistry of sediment core HE443/10-3, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.893766, 2018b.
Aromokeye, D. A., Kulkarni, A., Elvert, M., Wegener, G., Henkel, S., Coffinet, S., Eickhorst, T., Oni, O. E., Richter-Heitmann, T., Schnakenberg, A., Taubner, H., Wunder, L., Yin, X., Zhu, Q., Hinrichs, K.-U., Kasten, S., and Friedrich, M. W.: Rates and microbial players of iron-driven anaerobic oxidation of methane in methanic marine sediments, Front. Microbiol., 10, 3041, https://doi.org/10.3389/fmicb.2019.03041, 2020.
Aromokeye, D. A., Oni, O. E., Tebben, J., Yin, X., Richter-Heitmann, T., Wendt, J., Nimzyk, R., Littmann, S., Tienken, D., Kulkarni, A. C., Henkel, S., Hinrichs, K.-U., Elvert, M., Harder, T., Kasten, S., and Friedrich M. W.: Crystalline iron oxides stimulate methanogenic benzoate degradation in marine sediment-derived enrichment cultures, ISME J., 15, 965–980, https://doi.org/10.1038/s41396-020-00824-7, 2021.
Baloza, M., Henkel, S., Geibert, W., Kasten, S., and Holtappels, M.: Benthic carbon remineralization and iron cycling in relation to sea ice cover along the eastern continental shelf of the Antarctic Peninsula, J. Geophys. Res.-Oceans, 127, 7, e2021JC018401, https://doi.org/10.1029/2021JC018401, 2022.
Bao, P. and Li, G. X.: Sulfur-driven iron reduction coupled to anaerobic ammonium oxidation, Environ. Sci. Technol., 51, 6691–6698, https://doi.org/10.1021/acs.est.6b05971, 2017.
Beal, E. J., House, C. H., and Orphan, V. J.: Manganese- and iron-dependant marine methane oxidation, Science, 325,184-187, https://doi.org/10.1126/science.1169984, 2009.
Beard, B. L., Johnson, C. M., Cox, L., Sun, H., Nealson, K. H., and Aguliar, C.: Iron isotope biosignatures, Science, 285, 1889–1892, https://doi.org/10.1126/science.285.5435.1889, 1999.
Beard, B. L., Johnson, C. M., Skulan, J. L., Nealson, K. H., Cox, L., and Sun, H.: Application of Fe isotopes to tracing the geochemical and biological cycling of Fe, Chem. Geol., 195, 87–117, https://doi.org/10.1016/S0009-2541(02)00390-X, 2003a.
Beard, B. L., Johnson, C. M., Von Damm, K. L., and Poulson, R. L.: Iron isotope constraints on Fe cycling and mass balance in the oxygenated Earth oceans, Geology, 31, 629–632, https://doi.org/10.1130/0091-7613(2003)031<0629:IICOFC>2.0.CO;2, 2003b.
Beard, B. L., Handler, R. M., Scherer, M. M., Wu, L., Czaja, A. D., Heimann, A., and Johnson C. M.: Iron isotope fractionation between aqueous ferrous iron and goethite, Earth Planet. Sc. Lett., 295, 241–250, https://doi.org/10.1016/j.epsl.2010.04.006, 2010.
Beulig, F., Røy, H., Glombitza, C., and Jørgensen, B. B.: Control on rate and pathway of anaerobic organic carbon degradation in the seabed, P. Natl. Acad. Sci. USA, 115, 367–372, https://doi.org/10.1073/pnas.1715789115, 2018.
Bottrell, S. H., Parkes, R. J., Cragg, B. A., and Raiswell, R.: Isotopic evidence for anoxic pyrite oxidation and stimulation of bacterial sulphate reduction in marine sediments, J. Geol. Soc., 157, 711–714, https://doi.org/10.1144/jgs.157.4.711,2010.
Boudreau, B. P.: The diffusive tortuosity of fine-grained unlithified sediments, Geochim. Cosmochim. Ac., 60, 3139–3142, https://doi.org/10.1016/0016-7037(96)00158-5, 1996a.
Boudreau, B. P.: Diagenetic models and their implementation: Modelling transport and reactions in aquatic sediments, Springer, ISBN 13: 9783642643996, 1996b.
Burdige, D. J.: The biogeochemistry of manganese and iron reduction in marine sediments, Earth Sci. Rev., 35, 249–284, https://doi.org/10.1016/0012-8252(93)90040-E, 1993.
Butler, I. B., Archer, C., Vance, D., Oldroyd, A., and Rickard, D.: Fe isotope fractionation on FeS formation in ambient aqueous solution, Earth Planet. Sc. Lett., 236, 430–442, https://doi.org/10.1016/j.epsl.2005.05.022, 2005.
Canfield, D. E.: Reactive iron in marine sediments, Geochim. Cosmochim. Ac., 53, 619–632, https://doi.org/10.1016/0016-7037(89)90005-7, 1989.
Canfield, D. E., Raiswell, R., Westrich, J. T., Reaves, C. M., and Berner, R. A.: The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales, Chem. Geol., 54, 149–155, https://doi.org/10.1016/0009-2541(86)90078-1, 1986.
Chadwick, G. L., Nayak, D. D., Rother, M., and Metcalf, W. W.: No evidence for methanotrophic growth of diverse marine methanogens, P. Natl. Acad. Sci. USA, 121, 20, e2404143121, https://doi.org/10.1073/pnas.2404143121, 2024.
Cline, J. D.: Spectrophotometric determination of hydrogen sulfide in natural waters, Limnol. Oceanogr., 14, 454–458, https://doi.org/10.4319/lo.1969.14.3.0454, 1969.
Conway, T. M. and John, S. G.: Quantification of dissolved iron sources to the North Atlantic Ocean, Nature, 511, 212–215, https://doi.org/10.1038/nature13482, 2014.
Cornwell, J. C. and Morse, J. W.: The characterization of iron sulfide minerals in anoxic marine sediments, Mar. Chem., 22, 193–206, https://doi.org/10.1016/0304-4203(87)90008-9, 1987.
Crosby, H. A., Johnson, C. M., Roden, E. E., and Beard B. L.: Coupled Fe(II)-Fe(III) electron and atom exchange as a mechanism for Fe isotope fractionation during dissimilatory iron oxide reduction, Environ. Sci. Technol., 39, 6698–6704, https://doi.org/10.1021/es0505346, 2005.
Crosby, H. A., Roden, E. E., Johnson, C. M., and Beard, B. L.: The mechanism of iron isotope fractionation produced during dissimilatory Fe(III) reduction by Shewanella putrefaciens and Geobacter sulfurreducens, Geobiology, 5, 169–189, https://doi.org/10.1111/j.1472-4669.2007.00103.x, 2007.
Cruz Viggi, C., Rossetti, S., Fazi, S., Paiano, P., Majone, M., and Aulenta, F.: Magnetite particles triggering a faster and more robust syntrophic pathway of methanogenic propionate degradation, Environ. Sci. Technol., 48, 7536–43, https://doi.org/10.1021/es5016789, 2014.
D'Hondt, S., Jørgensen, B. B., Miller, D. J., Batzke, A., Blake, R., Cragg, B. A., Cypionka, H., Dickens, G. R., Ferdelman, T., Hinrichs, K.-U., Holm, N. G., Mitterer, R., Spivack, A., Wang, G., Bekins, B., Engelen, B., Ford, K., Gettemy, G., Rutherford, S. D., Sass, H., Skilbeck, C. G., Aiello, I. W., Guèrin, G., House, C. H., Inagaki, F., Meister, P., Naehr, T., Niitsuma, S., Parkes, R. J., Schippers, A., Smith, D. C., Teske, A., Wiegel, J., Padilla, C. N., and Acosta, J. L. S.: Distributions of microbial activities in deep subseafloor sediments, Science, 306, 2216–2221, https://doi.org/10.1126/science.1101155, 2004.
Egger, M., Rasigraf, O., Sapart, C. J., Jilbert, T., Jetten, M. S., Rockmann, T., van der Veen, C., Banda, N., Kartal, B., Ettwig, K. F., and Slomp, C. P.: Iron-mediated anaerobic oxidation of methane in brackish coastal sediments, Environ. Sci. Technol., 49, 277–283, https://doi.org/10.1021/es503663z, 2014.
Egger, M., Hagens, M., Sapart, C. J., Dijkstra, N., van Helmond, N. A. G. M., Mogollón, J. M., Risgaard-Petersen, N., van der Veen, C., Kasten, S., Riedinger, N., Böttcher, M. E., Rockmann, T., Jørgensen, B. B., and Slomp, C. P.: Iron oxide reduction in methane-rich deep Baltic Sea sediments, Geochim. Cosmochim. Ac., 207, 256–276, https://doi.org/10.1016/j.gca.2017.03.019, 2017.
Eliani-Russak, E., Tik, Z., Uzi-Gavrilov, S., Meijler, M. M., and Sivan, O.: The reduction of environmentally abundant iron oxides by the methanogen Methanosarcina barkeri, Front. Microbiol., 14, 1197299, https://doi.org/10.3389/fmicb.2023.1197299, 2023.
Findlay, A., Pellerin, A., Laufer, K., and Jørgensen, B. B.: Quantification of sulphide oxidation rates in marine sediments, Geochim. Cosmochim. Ac., 280, 441–452, 2020.
Fischer, D., Sahling, H., Nöthen, K., Bohrmann, G., Zabel, M., and Kasten, S.: Interaction between hydrocarbon seepage, chemosynthetic communities, and bottom water redox at cold seeps of the Makran accretionary prism: insights from habitat-specific pore water sampling and modeling, Biogeosciences, 9, 2013–2031, https://doi.org/10.5194/bg-9-2013-2012, 2012.
Gibson, J. and Harwood, S. C.: Metabolic diversity in aromatic compound utilization by anaerobic microbes, Annu. Rev. Microbiol., 56, 345–369, https://doi.org/10.1146/annurev.micro.56.012302.160749, 2012.
Gingele, F. X. and Kasten, S.: Solid-phase manganese in Southeast Atlantic sediments: Implications for the paleoenvironment, Mar. Geol., 121, 317–332, https://doi.org/10.1016/0025-3227(94)90037-X, 1994.
Glombitza, C., Egger, M., Røy, H., and Jørgensen, B. B.: Controls on volatile fatty acid concentrations in marine sediments (Baltic Sea), Geochim. Cosmochim. Ac., 258, 226–241, https://doi.org/10.1016/j.gca.2019.05.038, 2019.
Guilbaud, R., Butler, I. B., Ellam, R. M., and Rickard, D.: Fe isotope exchange between Fe(II)aq and nanoparticulate mackinawite (FeSm) during nanoparticle growth, Earth Planet. Sc. Lett., 300, 174–183, https://doi.org/10.1016/j.epsl.2010.10.004, 2010.
Guilbaud, R., Butler, I. B., and Ellam, R. M.: Abiotic pyrite formation produces a large Fe isotope fractionation, Science, 332, 1548–1551, https://doi.org/10.1126/science.1202924, 2011.
Gupta, D., Chen, K., Elliott, S. J., and Nayak, D. D.: MmcA is an electron conduit that facilitates both intracellular and extracellular electron transport in Methanosarcina acetivorans, Nat. Commun., 15, 3300, https://doi.org/10.1038/s41467-024-47564-2, 2024.
Hebbeln, D., Scheurle, C., and Lamy, F.: Depositional history of the Helgoland mud area, German Bight, North Sea, Geo-Mar. Lett., 23, 81–90, https://doi.org/10.1007/s00367-003-0127-0, 2003.
Henkel, S., Strasser, M., Schwenk, T., Hanebuth, T. J. J., Hüsener, J., Arnold, G. L., Winkelmann, D., Formolo, M., Tomasini, J., Krastel, S., and Kasten, S.: An interdisciplinary investigation of a recent submarine mass transport deposit at the continental margin off Uruguay, Geochem. Geophy. Geosy., 12, Q08009, https://doi.org/10.1029/2011GC003669, 2011.
Henkel, S., Kasten, S., Poulton, S. W., and Staubwasser, M.: Determination of the stable iron isotopic composition of sequentially leached iron phases in marine sediments, Chem. Geol., 421, 93–102, https://doi.org/10.1016/j.chemgeo.2015.12.003, 2016.
Henkel, S., Kasten, S., Hartmann, J. F., Silva-Busso, A., and Staubwasser, M.: Iron cycling and stable Fe isotope fractionation in Antarctic shelf sediments, King George Island, Geochim. Cosmochim. Ac., 237, 320–338, https://doi.org/10.1016/j.gca.2018.06.042, 2018.
Henkel, S., Staubwasser, M., Kasemann, S. A., Meixner, A., and Kasten, S.: Radiographs of the gravity core HE443/10-3 from North Sea, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.971531, 2024a.
Henkel, S., Staubwasser, M., Kasemann, S. A., Meixner, A., and Kasten, S.: Pore-water and solid-phase data of a multicorer core and a gravity core at Station HE443/10, North Sea, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.971490, 2024b.
Hensen, C., Zabel, M., Pfeifer, K., Schwenk, T., Kasten, S., Riedinger, N., Schulz, H. D., and Boetius, A.: Control of sulfate pore-water profiles by sedimentary events and the significance of anaerobic oxidation of methane for the burial of sulfur in marine sediments, Geochim. Cosmochim. Ac., 67:14, 2631–2647, https://doi.org/10.1016/S0016-7037(03)00199-6, 2003.
Hertweck, G.: Das Schlickgebiet in der inneren Deutschen Bucht. Aufnahme mit dem Sedimentechographen, Senck. Marit., 15, 219–249, 1983.
Heuer, V. B., Inagaki, F., Morono, Y., Kubo, Y., Maeda, L., Bowden, S., Cramm, M., Henkel, S., Hirose, T., Homola, K., Hoshino, T., Ijiri, A., Imachi, H., Kamiya, N., Kaneko, M., Lagostina, L., Manners, H., McClelland, H. L., Metcalfe, K., Okutsu, N., Pan, D., Raudsepp, M. J., Sauvage, J., Schubotz, F., Spivack, A., Tonai, S., Treude, T., Tsang, M. Y., Viehweger, B., Wang, D. T., Whitaker, E., Yamamoto, Y., Yang, K., and Kinoshita, M.: Expedition 370 Preliminary Report: Temperature Limit of the Deep Biosphere off Muroto, International Ocean Discovery Program, https://doi.org/10.14379/iodp.pr.370.2017, 2017.
Holmkvist, L., Ferdelman, T. G., and Jørgensen, B. B.: A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark), Geochim. Cosmochim. Ac., 75, 3581–3599, https://doi.org/10.1016/j.gca.2011.03.033, 2011.
Homoky, W. B., Severmann, S., Mills, R. A., Statham, P. J., and Fones, G. R.: Pore-fluid Fe isotopes reflect the extent of benthic Fe redox recycling: Evidence from continental shelf and deep-sea sediments, Geology, 37, 751–754, https://doi.org/10.1130/G25731A.1, 2009.
Hopkins, B. T., McInerney, M. J., and Warikoo, V.: Evidence for anaerobic syntrophic benzoate degradation threshold and isolation of the syntrophic benzoate degrader, Appl. Environ. Microb., 61, https://doi.org/10.1128/aem.61.2.526-530.1995, 1995.
Ionescu, D., Heim, C., Polerecky, L., Thiel, V., and De Beer, D.: Biotic and abiotic oxidation and reduction of iron at circumneutral pH are inseparable processes under natural conditions, Geomicrobiol. J., 32:3-4, 221–230, https://doi.org/10.1080/01490451.2014.887393, 2015.
Irion, G., Wunderlich, F., and Schwedhelm, E.: Transport of clay-minerals and anthropogenic compounds into the German Bight and the provenance of fine-grained sediments SE of Helgoland, J. Geol. Soc. London, 144, 153–160, https://doi.org/10.1144/gsjgs.144.1.0153, 1987.
Jiang, S., Park, S., Yoon, Y., Lee, J.-H., Wu, M.-W., Phuoc, Dan N., Sadowsky, M. J., and Hur, H.-G.: Methanogenesis facilitated by geobiochemical iron cycle in a novel syntrophic methanogenic microbial community, Environ. Sci. Technol., 47, 10078–10084, https://doi.org/10.1021/es402412c, 2013.
John, S. G., Mendez, J., Moffett, J., and Adkins, J.: The flux of iron and iron isotopes from San Pedro Basin sediments, Geochim. Cosmochim. Ac., 93, 14–29, https://doi.org/10.1016/j.gca.2012.06.003, 2012.
Johnson, C. M. and Beard B. L.: Biogeochemical cycling of iron isotopes, Science, 309, 1025–1027, https://doi.org/10.1126/science.1112552, 2005.
Johnson, C. M., Beard, B. L., Roden, E. E., Newman, D. K., and Nealson, K. H.: Isotopic constraints on biogeochemical cycling of Fe, Rev. Mineral. Geochem., 55, 359–408, https://doi.org/10.2138/gsrmg.55.1.359, 2004.
Johnson, C. M., Roden, E. E., Welch, S. A., and Beard, B. L.: Experimental constraints on Fe isotope fractionation during magnetite and Fe carbonate formation coupled to dissimilatory hydrous ferric oxide reduction, Geochim. Cosmochim. Ac., 69, 963–993, https://doi.org/10.1016/j.gca.2004.06.043, 2005.
Jørgensen, B. B.: Bacteria and Marine Biogeochemistry, in: Marine Geochemistry, edited by: Schulz, H. D. and Zabel, M., Springer Berlin Heidelberg, Berlin, Heidelberg, https://doi.org/10.1007/978-3-662-04242-7_5, pp. 169–206, 2006.
Jørgensen, B. B. and Kasten, S.: Sulfur cycling and methane oxidation, in: Marine Geochemistry, edited by: Schulz H. D. and Zabel M., Springer-Verlag, Heidelberg, https://doi.org/10.1007/3-540-32144-6_8, 574, 2006.
Kasten, S., Freudenthal, T., Gingele, F. X., and Schulz, H. D.: Simultaneous formation of iron-rich layers at different redox boundaries in sediments of the Amazon deep-sea fan, Geochim. Cosmochim. Ac., 62:13, 2253–2264, https://doi.org/10.1016/S0016-7037(98)00093-3, 1998.
Kato, S., Hashimoto, K., and Watanabe, K.: Methanogenesis facilitated by electric syntrophy via (semi) conductive iron-oxide minerals, Environ. Microbiol., 14, 1646–1654, https://doi.org/10.1111/j.1462-2920.2011.02611.x, 2012.
Keeling, C. D.: The concentration and isotopic abundances of atmospheric carbon dioxide in rural areas, Geochim. Cosmochim. Ac., 13, 322–334, https://doi.org/10.1016/0016-7037(58)90033-4, 1958.
Kerouel, R. and Aminot, A.: Fluorimetric determination of ammonia in sea and estuarine waters by direct segmented flow analysis, Mar. Chem., 57, 3–4, 265–275, https://doi.org/10.1016/S0304-4203(97)00040-6, 1997.
Köster, M., Staubwasser, M., Meixner, A., Kasemann, S. A., Manners, H. R., Morono, Y., Inagaki, F., Heuer, V. B., Kasten, S., and Henkel, S.: Uniquely low stable iron isotopic signatures in deep marine sediments caused by Rayleigh distillation, Sci. Rep., 13, 10281, https://doi.org/10.1038/s41598-023-37254-2, 2023.
Larese-Casanova, P., Yue, P., and Gorski, C.: Fe and O atom exchange on hematite and goethite surfaces after aqueous Fe(II) sorption: A NanoSIMS investigation, Goldschmidt 2023 Abstract, https://doi.org/10.7185/gold2023.16329, 2023.
LaRowe, D. E. and Van Capellen, P.: Degradation of natural organic matter: A thermodynamic analysis, Geochim. Cosmochim. Ac., 75, 2030–2042, https://doi.org/10.1016/j.gca.2011.01.020, 2011.
Lehours, A. C., Rabiet, M., Morel-Desrosiers, N., Morel, J.-P., Jouve, L., Arbeille, B., Mailhot, G., and Fonty, G.: Ferric iron reduction by fermentative strain BS2 isolated from an iron-rich anoxic environment (Lake Pavin, France), Geomicrobiol. J., 27, 714–722, https://doi.org/10.1080/01490451003597663, 2010.
Lovley, D. R.: Dissimilatory Fe(III) and Mn(IV) reduction, Microbiol. Rev., 55, 259–287, https://doi.org/10.1016/S0065-2911(04)49005-5, 1991.
Lovley, D. R. and Holmes, D. E.: Electromicrobiology: the ecophysiology of phylogenetically diverse electroactive microorganisms, Nat. Rev. Microbiol., 20, 6–19, https://doi.org/10.1038/s41579-021-00597-6, 2022.
Lovley, D. R. and Phillips, E. J.: Organic matter mineralization with reduction of ferric iron in anaerobic sediments, Appl. Environ. Microb., 51, 683–689, https://doi.org/10.1128/aem.51.4.683-689.1986,1986.
Lovley, D. R., Phillips, E. J. P., and Lonergan, D. J.: Hydrogen and formate oxidation coupled to dissimilatory reduction of iron or manganese by Alteromonas-putrefaciens, Appl. Environ. Microb., 55, 700–706, https://doi.org/10.1128/aem.55.3.700-706.1989, 1989.
März, C., Hoffmann, J., Bleil, U., De Lange, G. J., and Kasten, S.: Diagenetic changes of magnetic and geochemical signals by anaerobic methane oxidation in sediments of the Zambezi deep-sea fan (SW Indian Ocean), Mar. Geol., 255:3–4, 118–130, https://doi.org/10.1016/j.margeo.2008.05.013, 2008.
Michaud, A. B., Laufer, K., Findlay, A., Pellerin, A., Antler, G., Turchyn, A. V., Røy, H., Wehrmann, L. M., and Jørgensen, B. B.: Glacial influence on the iron and sulfur cycles in Arctic fjord sediments (Svalbard), Geochim. Cosmochim. Ac., 280, 423–440, https://doi.org/10.1016/j.gca.2019.12.033, 2020.
Miller, J. B. and Tans, P. P.: Calculating isotopic fractionation from atmospheric measurements at various scales, Tellus B, 55, 207–214, https://doi.org/10.1034/j.1600-0889.2003.00020.x, 2003.
Mortimer, R. J. G., Galsworthy, A. M. J., Bottrell, S. H., Wilmot, L. E., and Newton, R. J.: Experimental evidence for rapid biotic and abiotic reduction of Fe(III) at low temperatures in salt marsh sediments: a possible mechanism for formation of modern sedimentary siderite concretions, Sedimentology, 58, 1514–1529, https://doi.org/10.1111/j.1365-3091.2011.01224.x, 2011.
Oni, O., Miyatake, T., Kasten, S., Richter-Heitmann, T., Fischer, D., Wagenknecht, L., Kulkarni, A., Blumers, M., Shylin, S. I., Ksenofontov, V., Costa, B. F. O., Klingelhöfer, G., and Friedrich, M. W.: Distinct microbial populations are tightly linked to the profile of dissolved iron in the methanic sediments of the Helgoland mud area, North Sea, Front. Microbiol., 6, https://doi.org/10.3389/fmicb.2015.00365, 2015a.
Oni, E. O., Schmidt, F., Miyatake, T., Kasten, S., Witt, M., Hinrichs, K.-U., and Friedrich, M.: Microbial communities and organic matter composition in surface and subsurface sediments of the Helgoland mud area, North Sea, Front. Microbiol., 6, 1290, https://doi.org/10.3389/fmicb.2015.01290, 2015b.
Parkhurst, D. L. and Appelo, C. A. J.: Description of input and examples for PHREEQC version 3 – a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations, in: U. S. Geological Survey Techniques and Methods, 6, U.S. Geological Survey, Reston, USA, https://doi.org/10.3133/tm6A43, 2013.
Pataki, D. E., Ehleringer, J. R., Flanagan, L. B., Yakir, D., Bowling, D. R., Still, C. J., Buchmann N., Kaplan, J. O., and Berry J. A.: The application and interpretation of Keeling plots in terrestrial carbon cycle research, Global Biogeochem. Cy., 17, 1–14, https://doi.org/10.1029/2001GB001850, 2013.
Poulton, S. W. and Canfield, D. E.: Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates, Chem. Geol., 214, 209–221, https://doi.org/10.1016/j.chemgeo.2004.09.003, 2005.
Poulton, S. W., Krom, M. D., and Raiswell, R.: A revised scheme for the reactivity of iron (oxyhydr)oxides towards dissolved sulfide, Geochim. Cosmochim. Ac., 68, 3703–3715, https://doi.org/10.1016/j.gca.2004.03.012, 2004.
Praharaj, T. and Fortin, D.: Determination of acid volatile sulfides and chromium reducible sulfides in Cu-Zn and Au mine tailings, Water Air Soil Poll, 155, 35–50, https://doi.org/10.1023/B:WATE.0000026526.26339.c3, 2003.
Pyzik, A. J. and Sommer, S. E.: Sedimentary iron monosulfides: kinetics and mechanism of formation, Geochim. Cosmochim. Ac., 45, 687–698, https://doi.org/10.1016/0016-7037(81)90042-9, 1981.
Reed, D. C., Slomp C. P., and Gustafsson, B. G.: Sedimentary phosphorus dynamics and the evolution of bottom-water hypoxia: A coupled benthic-pelagic model of a coastal system, Limnol. Oceanogr., 56, 1075–1092, https://doi.org/10.4319/lo.2011.56.3.1075, 2011a.
Reed, D. C., Slomp, C. P., and de Lange, G. J.: A quantitative reconstruction of organic matter and nutrient diagenesis in Mediterranean Sea sediments over the Holocene, Geochim. Cosmochim. Ac., 75, 5540–5558, https://doi.org/10.1016/j.gca.2011.07.002, 2011b.
Riedinger, N., Pfeifer, K., Kasten, S., Garming, J. F. L., Vogt, C., and Hensen, C.: Diagenetic alteration of magnetic signals by anaerobic oxidation of methane related to a change in sedimentation rate, Geochim. Cosmochim. Ac., 69, 4117–4126, https://doi.org/10.1016/j.gca.2005.02.004, 2005.
Riedinger, N., Brunner, B., Formolo, M. J., Solomon, E., Kasten, S., Strasser, M., and Ferdelman, T. G.: Oxidative sulfur cycling in the deep biosphere of the Nankai Trough, Japan, Geology, 38, 851–854, https://doi.org/10.1130/G31085.1, 2010.
Riedinger, N., Formolo, M. J., Lyons, T. W., Henkel, S., Beck, A., and Kasten, S.: An inorganic geochemical argument for coupled anaerobic oxidation of methane and iron reduction in marine sediments, Geobiology, 12, 172–181, https://doi.org/10.1111/gbi.12077, 2014.
Riedinger, N., Brunner, B., Krastel, S., Arnold, G. L., Wehrmann, L. M., Formolo, M. J., Beck, A., Bates, S. M., Henkel, S., Kasten, S., and Lyons, T. W.: Sulfur cycling in an iron oxide-dominated, dynamic marine depositional system: The Argentine continental margin, Front. Earth Sci., 5, 33, https://doi.org/10.3389/feart.2017.00033, 2017.
Roden, E. E. and Lovley, D. R.: Dissimilatory Fe(III) reduction by the marine microorganism Desulfuromonas acetoxidans, Appl. Environ. Microb., 59, 734–742, https://doi.org/10.1128/aem.59.3.734-742.1993, 1993.
Rothe, M., Frederichs, T., Eder, M., Kleeberg, A., and Hupfer, M.: Evidence for vivianite formation and its contribution to long-term phosphorus retention in a recent lake sediment: a novel analytical approach, Biogeosciences, 11, 5169–5180, https://doi.org/10.5194/bg-11-5169-2014, 2014.
Roy, M., Rouxel, O., Martin, J. B., and Cable, J. E.: Iron isotope fractionation in a sulfide-bearing subterranean estuary and its potential influence on oceanic Fe isotope flux, Chem. Geol., 300–301, 133–142, https://doi.org/10.1016/j.chemgeo.2012.01.022, 2012.
Rullkötter, J.: Organic Matter: The driving force for early diagenesis, in: Marine Geochemistry, edited by: Schulz H. D. and Zabel M., Springer, Berlin, Heidelberg, https://doi.org/10.1007/3-540-32144-6_4, 2006.
Schoenberg, R. and von Blanckenburg, F.: An assessment of the accuracy of stable Fe isotope ratio measurements on samples with organic and inorganic matrices by high-resolution multicollector ICP-MS, Int. J. Mass Spectrom., 242, 257–272, https://doi.org/10.1016/j.ijms.2004.11.025, 2005.
Scholz, F., Severmann, S., McManus, J., Noffke, A., Lomnitz, U., and Hensen, C.: On the isotope composition of reactive iron in marine sediments: Redox shuttle versus early diagenesis, Chem. Geol., 389, 48–59, https://doi.org/10.1016/j.chemgeo.2014.09.009, 2014.
Shah, M., Lin, C.-C., Kukkadapu, R., Engelhard, M. H., Zhao, X., Wang, Y., Barkay, T., and Yee, N.: Syntrophic Effects in a Subsurface Clostridial Consortium on Fe(III)-(Oxyhydr)oxide Reduction and Secondary Mineralization, Geomicrobiol. J., 31, 101–115.https://doi.org/10.1080/01490451.2013.806610, 2013.
Seeberg-Elverfeldt, J., Schlüter, M., Feseker, T., and Kölling, M.: Rhizon sampling of porewaters near the sediment-water interface of aquatic systems, Limnol. Oceanogr.-Meth., 3, 361–371, https://doi.org/10.4319/lom.2005.3.361, 2005.
Segarra, K. E. A., Comerford, C., Slaughter, J., and Joye, S. B.: Impact of electron acceptor availability on the anaerobic oxidation of methane in coastal freshwater and brackish wetland sediments, Geochim. Cosmochim. Ac., 115, 15–30, https://doi.org/10.1016/j.gca.2013.03.029, 2013.
Severmann, S., Johnson, C. M., Beard, B. L., and McManus, J.: The effect of early diagenesis on the Fe isotope compositions of porewaters and authigenic minerals in continental margin sediments, Geochim. Cosmochim. Ac., 70, 2006–2022, https://doi.org/10.1016/j.gca.2006.01.007, 2006.
Severmann, S., McManus, J., Berelson, W. M., and Hammond, D. E.: The continental shelf benthic iron flux and its isotope composition, Geochim. Cosmochim. Ac., 74, 3984–4004, https://doi.org/10.1016/j.gca.2010.04.022, 2010.
Sieber, M., Conway, T. M., De Souza, G. F., Hassler, C. S., Ellwood, M. J., and Vance, D.: Isotopic fingerprinting of biogeochemical processes and iron sources in the iron-limited surface Southern Ocean, Earth Planet. Sc. Lett., 567, 116967, https://doi.org/10.1016/j.epsl.2021.116967, 2021.
Sivan, O., Adler, M., Pearson, A., Gelman, F., Bar-Or, I., John, S. G., and Eckert, W.: Geochemical evidence for iron-mediated anaerobic oxidation of methane, Limnol. Oceanogr., 56, 1536–1544, https://doi.org/10.4319/lo.2011.56.4.1536, 2011.
Sivan, O., Shusta, S. S., and Valentine, L.: Methanogens rapidly transition from methane production to iron reduction, Geobiology, 14:2, 190–203, https://doi.org/10.1111/gbi.12172, 2016.
Staubwasser, M., von Blanckenburg, F., and Schoenberg, R.: Iron isotopes in the early marine diagenetic iron cycle, Geology, 34, 629–632, https://doi.org/10.1130/G22647.1, 2006.
Stoll, M. H. C., Bakker, K., Nobbe, G. H., and Haese, R. R.: Continuous-flow analysis of dissolved inorganic carbon content in seawater, Anal. Chem., 73, 4111–4116, https://doi.org/10.1021/ac010303r, 2001.
Stookey, L. L.: Ferrozine – A new spectrophotometric reagent for iron, Anal. Chem., 42, 779–781, https://doi.org/10.1021/ac60289a016, 1970.
Tessier, A., Campbell, P. G. C., and Bisson, M.: Sequential extraction procedure for the speciation of particulate trace metals, Anal. Chem., 51, 844–851, https://doi.org/10.1021/ac50043a017, 1979.
Thamdrup, B., Finster, K., Würgler, Hansen J., and Bak, F.: Bacterial disproportionation of elemental sulfur coupled to chemical reduction of iron or manganese, Appl. Environ. Microb., 59, 101–108, https://doi.org/10.1128/aem.59.1.101-108.1993, 1993.
Thamdrup, B., Fossing, H., and Jørgensen, B. B.: Manganese, iron, and sulfur cycling in a coastal marine sediment, Aarhus Bay, Denmark, Geochim. Cosmochim. Ac., 58, 5115–5129, https://doi.org/10.1016/0016-7037(94)90298-4, 1994.
Thiel, J., Byrne, J. M., Kappler, A., Schink, B., and Pester, M.: Pyrite formation from FeS and H2S is mediated through microbial redox activity, P. Natl. Acad. Sci. USA, 116, 6897–6902, https://doi.org/10.1073/pnas.1814412116, 2019.
Vuillemin, A., Friese, A., Wirth, R., Schuessler, J. A., Schleicher, A. M., Kemnitz, H., Lücke, A., Bauer, K. W., Nomosatryo, S., von Blanckenburg, F., Simister, R., Ordoñez, L. G., Ariztegui, D., Henny, C., Russell, J. M., Bijaksana, S., Vogel, H., Crowe, S. A., Kallmeyer, J., and the Towuti Drilling Project Science team: Vivianite formation in ferruginous sediments from Lake Towuti, Indonesia, Biogeosciences, 17, 1955–1973, https://doi.org/10.5194/bg-17-1955-2020, 2020.
Walczyk, T. and von Blanckenburg, F.: Deciphering the iron isotope message of the human body, Int. J. Mass Spec., 242, 117–134, https://doi.org/10.1016/j.ijms.2004.12.028, 2005.
Wang, Y. and Van Capellen, P.: A multicomponent reactive transport model of early diagenesis: Application to redox cycling in coastal marine sediments, Geochim. Cosmochim. Ac., 60, 2993–3014, https://doi.org/10.1016/0016-7037(96)00140-8, 1996.
Wedepohl, K. H.: Chemical composition and fractionation of the continental crust, Geol. Rundsch., 80, 207–223, https://doi.org/10.1007/BF01829361, 1991.
Wersin, P., Höhener, P., Giovanoli, R., and Stumm, W.: Early diagenetic influences on iron transformations in a freshwater lake sediment, Chem. Geol., 90, 233–252, https://doi.org/10.1016/0009-2541(91)90102-W, 1991.
Weyer, S. and Schwieters, J. B.: High precision Fe isotope measurements with high mass resolution MC-ICPMS, Int. J. Mass Spectrom., 226, 355–368, https://doi.org/10.1016/S1387-3806(03)00078-2, 2003.
Whiticar, M. J.: Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane, Chem. Geol., 161, 291–314, https://doi.org/10.1016/S0009-2541(99)00092-3, 1999.
Wieder, R. K., Lang, G. E., and Granus, V. A.: An evaluation of wet chemical methods for quantifying sulfur fractions in freshwater wetland peat, Limnol. Oceanogr., 30, 1109–1115, https://doi.org/10.4319/lo.1985.30.5.1109, 1985.
Wiederhold, J. G.: Metal stable isotope signatures as tracers in environmental geochemistry, Environ. Sci. Technol., 49, 2606–2624, https://doi.org/10.1021/es504683e, 2015.
Wiesli, R. A., Beard, B. L., and Johnson, C. M.: Experimental determination of Fe isotope fractionation between aqueous Fe(II), siderite and “green rust” in abiotic systems, Chem. Geol., 211, 343–362, https://doi.org/10.1016/j.chemgeo.2004.07.002, 2004.
Wunder, L. C., Breuer, I., Willis-Poratti, G., Aromokeye, D. A., Henkel, S., Richter-Heitmann, T., Yin, X., Friedrich, M. W.: Manganese reduction and microbial communities involved in Antarctic surface sediments from Potter Cove, Front. Microbiol., 15, 1398021, https://doi.org/10.3389/fmicb.2024.1398021, 2024.
Xu, H., Chang, J., Wang, H., Liu, Y., Zhang, X., Liang, P., and Huang, X.: Enhancing direct interspecies electron transfer in syntrophic-methanogenic associations with (semi)conductive iron oxides: Effects and mechanisms, Sci. Total Environ., 695, 133876, https://doi.org/10.1016/j.scitotenv.2019.133876, 2019.
Yin, X., Zhou, G., Wang, H., Han, D., Maeke, M., Richter-Heitmann, T., Aromokeye, D., Zhu, Q., Nimzyk, R., Elvert, M., and Friedrich, M. W.: Unexpected carbon utilization activity of sulfate-reducing microorganisms in temperate and permanently cold marine sediments, ISME J., 18, 1–14, https://doi.org/10.1093/ismejo/wrad014, 2024.
Yu, L., He, D., Yang, L., Rensing, C., Zeng, R. J., and Zhou, S.: Anaerobic methane oxidation coupled to ferrihydrite reduction by Methanosarcina barkeri, Sci. Total Environ., 844, 157235, https://doi.org/10.1016/j.scitotenv.2022.157235, 2022.
Zhou, Z., Henkel, S., Kasten, S., and Holtappels, M.: The iron “redox battery” in sandy sediments: Its impact on organic matter remineralization and phosphorus cycling, Sci. Total Environ., 865, 161168, https://doi.org/10.1016/j.scitotenv.2022.161168, 2023.
Zhou, Z., Waska, H., Henkel, S., Dittmar, T., Kasten, S., and Holtappels, M.: Iron promotes the retention of terrigenous dissolved organic matter in subtidal permeable sediments, Environ. Sci. Technol., 58, 14, 6204–6214, https://doi.org/10.1021/acs.est.3c09531, 2024.
Zhuang, L., Tang, J., Wang, Y., Hu, M., and Zhou, S.: Conductive iron oxide minerals accelerate syntrophic cooperation in methanogenic benzoate degradation, J. Hazard. Mater., 293, 37–45, https://doi.org/10.1016/j.jhazmat.2015.03.039, 2015.