the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 Apr 2020
| 03 Apr 2020
Oxygen isotope composition of waters recorded in carbonates in strong clumped and oxygen isotopic disequilibrium
Caroline Thaler
Amandine Katz
Magali Bonifacie
Bénédicte Ménez
Magali Ader
Paleoenvironmental reconstructions, which are mainly retrieved from oxygen isotope (δ18O) and clumped isotope (Δ47) compositions of carbonate minerals, are compromised when carbonate precipitation occurs in isotopic disequilibrium. To date, knowledge of these common isotopic disequilibria, known as vital effects in biogenic carbonates, remains limited, and the potential information recorded by δ18O and Δ47 offsets from isotopic equilibrium values is largely overlooked. Additionally, in carbonates formed in isotopic equilibrium, the use of the carbonate δ18O signature as a paleothermometer relies on our knowledge of the paleowaters' δ18O value, which is often assumed. Here, we report the largest Δ47 offsets observed to date (as much as −0.270 ‰), measured on microbial carbonates that are strongly linked to carbonate δ18O offsets (−25 ‰) from equilibrium. These offsets are likely both related to the microorganism metabolic activity and yield identical erroneous temperature reconstructions. Unexpectedly, we show that the δ18O value of the water in which carbonates precipitated, as well as the water–carbonate δ18O fractionation dependence on temperature at equilibrium, can be retrieved from these paired δ18O and Δ47 disequilibrium values measured in carbonates. The possibility to retrieve the δ18O value of paleowaters, sediments' interstitial waters or organisms' body water at the carbonate precipitation loci, even from carbonates formed in isotopic disequilibrium, opens long-awaited research avenues for both paleoenvironmental reconstructions and biomineralization studies.
- Article
(1448 KB) - Full-text XML
-
Supplement
(481 KB) - BibTeX
- EndNote





Oxygen isotope composition (δ18O) paired with clumped isotope composition (Δ47) of carbonate minerals is increasingly used for reconstructing paleoenvironmental or diagenetic conditions (Ghosh et al., 2006; Henkes et al., 2018; Mangenot et al., 2018a, b). The δ18O of carbonates depends on both the δ18O value of the water in which the carbonate precipitated and the precipitation temperature (Urey et al., 1951). Its use to reconstruct paleoenvironments can be combined with the new carbonate C–O “clumped isotope” abundance (Δ47) thermometer which depends only on the carbonate precipitation temperature (Ghosh et al., 2006). By combining the Δ47-derived temperatures and the carbonate δ18O value (δ18Ocarbonate), the δ18O value of the water (δ18Owater) in which the carbonate precipitated can be retrieved. However, this requires that solid carbonate and water reached isotopic equilibrium, which is often hard to prove. Conversely, carbonate precipitation in isotopic disequilibrium is commonly encountered (Affek et al., 2014; Loyd et al., 2016). Out of equilibrium, δ18O and Δ47 values are particularly known to occur in biogenic carbonates (Thiagarajan et al., 2011; Bajnai et al., 2018) – the most abundant carbonates in the sedimentary record. To date, the reasons for these isotopic disequilibria in carbonates remain largely under-constrained. While Δ47 compositions of carbonates seemed at first free of any biologically driven or mineral-specific fractionation known to affect δ18Ocarbonate compositions (Eiler, 2011), recently identified disequilibrium Δ47 values (Saenger et al., 2012; Affek, 2013; Tang et al., 2014; Burgener et al., 2018) shed new light on and help to unravel the mechanisms responsible for oxygen isotopic disequilibrium in carbonate minerals. Even though the identification of these vital effects does not prevent δ18O and Δ47 tools from being powerful paleothermometers as empirical calibrations taking vital effects into account allow temperature reconstructions, it has become crucial to determine if the δ18O and Δ47 disequilibria observed in carbonates as diverse as those found in coral reefs (Saenger et al., 2012), brachiopods (Bajnai et al., 2018), microbialites and methane seep carbonates (Loyd et al., 2016) along with speleothems (Affek et al., 2014) could be explained by oxygen isotope disequilibria occurring in dissolved inorganic carbon (DIC) involved in carbonate precipitation. In this case, δ18O and Δ47 disequilibria in biogenic carbonates would record information, however unavailable yet, on the physiological characteristics of carbonate-forming organisms.
In previous experiments we produced microbial calcium carbonates (Millo et al., 2012; Thaler et al., 2017) that recorded the strongest oxygen isotope disequilibrium ever identified between DIC and precipitation water (i.e., −25 ‰ offset from δ18Ocarbonate equilibrium values). We used carbonic anhydrase (CA), an enzyme able to accelerate oxygen isotope equilibration between DIC and water via fast CO2 hydration and dehydration. When CA was added to the precipitation water, the carbonate oxygen isotope compositions reached equilibrium with the precipitation water (Thaler et al., 2017). Here, we build on these experiments as they offer a unique opportunity to assess experimentally whether carbonates precipitated from DIC in disequilibrium with water also record Δ47 disequilibrium values and the type of information that is actually carried by these paired disequilibria. We later show how and to what extent this can be applied to previously published cases of oxygen isotopic offsets from equilibrium values in both biogenic and abiotic carbonates.
2.1 Precipitation of microbial carbonates
Carbonates were precipitated at 30±0.1 ∘C using the procedure detailed in Millo et al. (2012) and Thaler et al. (2017) and summarized hereafter. The precipitation solution (initial pH = 6.0) was composed of ions added to Milli-Q® water (resistivity = 18 MΩ cm) by dissolving salts in the following order: MgSO4•7H2O (16 mM), NaCl (80 mM), KCl (4 mM), urea (33.3 mM) and CaCl2 (40 mM). The aim was to mimic the ionic composition of groundwater (Millo et al., 2012). In experiments with CA whose δ18O results (but not the Δ47 ones) were recently published in Thaler et al. (2017), the precipitation solution was supplemented with CA at a concentration of 2 mg L−1. The precipitation solution (with or without CA) was then mixed at a volumetric ratio of 1 : 1 with the ureolytic soil bacteria Sporosarcina pasteurii (Fig. 1) suspended in Milli-Q® water, at a final optical density at 600 nm of 0.100±0.010. For this study, 16 gastight Exetainer® vials were filled with the precipitation solution without CA in order to sacrifice them at regular time intervals (i.e., 30, 60, 120, 180 and 360 min and 24 h) and thus obtain information on the kinetics of the reaction, while reproducing the procedure followed for the experiment with CA (Thaler et al., 2017) consisting of 27 vials sacrificed every 10 to 30 min and after 24 h. The vials capped with rubber septa were filled up to the brim, i.e., without headspace, hence preventing any gaseous exchange with the atmosphere or headspace gases.

Figure 1Scanning electron microscopy images of the bio-induced carbonates formed with CA (almost pure calcite and traces of vaterite and aragonite as determined using X-ray diffraction; Thaler et al., 2017). The fingerprints of Sporosarcina pasteurii cells are visible as black holes (a showing a cross section of carbonate) or as rods and indicated by white arrows.
Ureolysis corresponds to two types of hydrolysis: (i) the hydrolysis of urea into ammonia (NH3) and carbamate (H2N-COOH) (H2N-CO-NH2 + H2O → NH3 + H2N-COOH), which is catalyzed by urease and is the rate-limiting step, and (ii) the rapid and spontaneous hydrolysis of carbamate into ammonia and CO2(aq) (H2N-COOH + H2O → NH3 + CO2(aq) + H2O) (Krebs and Roughton, 1948; Matsuzaki et al., 2013) or H2CO3 (H2N-COOH + H2O → NH3 + H2CO3) (Mobley and Hausinger, 1989; Krajewska, 2009).
Ureolysis completion was followed by evaluating the production of dissolved inorganic nitrogen (DIN = NH3 + ). Determination of pH, DIN concentration and amount of precipitated carbonates (Fig. 2), as well as isotopic measurements, was performed for each vial to monitor their evolution as the ureolysis reaction progresses. The pH initially increased from 6.0 to 9.0 due to NH3 production by ureolysis and consecutive alkalinization of the precipitation solution (Fig. 2a). The subsequent carbonate precipitation (Fig. 2b) lowered pH to 8.6 (without CA) and 8.4 (with CA) and was followed by a second pH increase to 8.8 (without CA) and 8.7 (with CA) when carbonate precipitation stopped while ureolysis continued. At ureolysis completion, all the calcium initially present in solution (i.e., the limiting reagent) had precipitated whereas 35 % to 45 % of the DIC produced by ureolysis remained in solution. Carbonate precipitates, formed at the bottom and on the wall of the vials, were immediately rinsed with a few drops of pure ethanol in order to dehydrate bacteria and prevent further ureolysis, carbonate formation and/or dissolution–reprecipitation processes. Ethanol was then removed, and, prior to their collection, carbonates were dried overnight at 40 ∘C in the vials placed in a ventilated oven equipped with desiccating beads. The majority of the carbonates precipitated in this study were composed of calcite with minor amounts of aragonite (1 % to 4 %), vaterite (2 % to 4 %) and magnesian calcite with low Mg content (Mg0.064, Ca0.936)CO3 (up to 8 %) (Thaler et al., 2017).
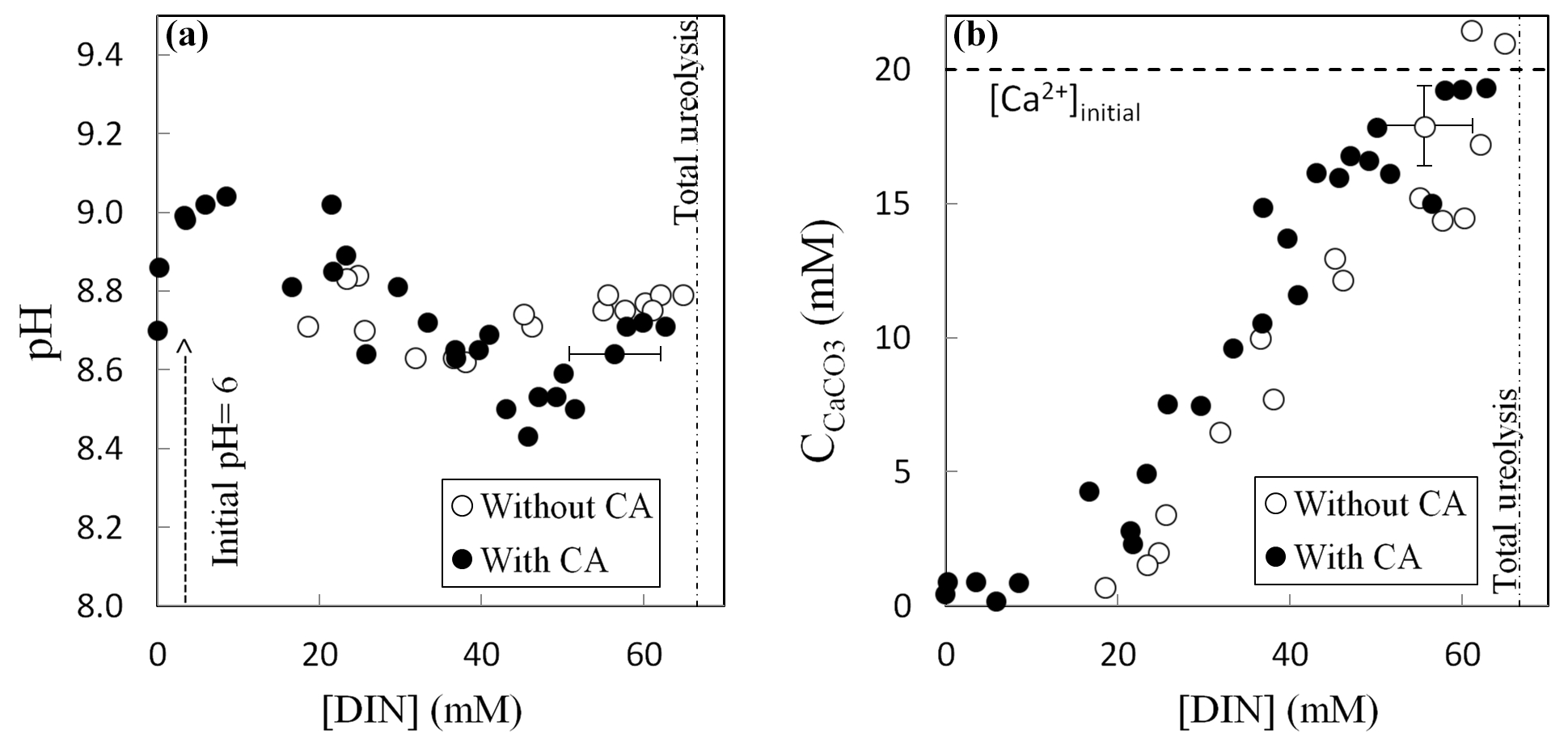
Figure 2Evolution of pH (a) and amount of precipitated calcium carbonate (b) as a function of the production of dissolved inorganic nitrogen DIN () by bacteria during ureolysis, without carbonic anhydrase (CA) (this study) and with CA (data from Thaler et al., 2017). Error bars account for 2 SD and are always smaller than symbols for pH values. The mineralogy of the carbonates precipitated with or without CA was determined by X-ray diffraction, indicating that solid carbonates are mostly composed of calcite (including up to 8 % of low-Mg calcite, (Mg0.064, Ca0.936)CO3), vaterite (2 %–4 %) and aragonite (1 %–4 %) (Thaler et al., 2017). The influence of mineralogy on isotopic measurements and temperature estimates is discussed in the Supplement, Sect. S1.
All of the measured chemical parameters (pH, DIC, amount of solid carbonates, Ca2+ concentration, DIN) along with DIC and solid carbonate δ13C behave similarly with or without active CA (Thaler et al., 2017). It was not possible to measure Δ47 for all the precipitated carbonates due to their low amount, particularly for the tubes sacrificed at the beginning of the experiments (Table S1 in the Supplement).
2.2 δ18O and Δ47 measurements and associated uncertainties
All the isotopic analyses were made at Institut de physique du globe de Paris (IPGP, France). δ18O analyses were performed on carbonate powders of ca. 2 mg with a continuous-helium-flow isotope ratio mass spectrometer AP 2003 (Analytical Precision 2003, GV Instruments) coupled to a gas chromatograph column (GC-IRMS, Chrompac Column Type 99960), as described in Millo et al. (2012) and Thaler et al. (2017). External reproducibility on carbonate standards is ±0.1 ‰ (1 SD) and represents the uncertainty assigned to δ18Ocarbonate data.
The analytical procedure used for clumped isotope Δ47 measurements is only briefly presented here and detailed in Bonifacie et al. (2017). About 5 mg of carbonates was digested at 90 ∘C for 20 min with 104 % phosphoric acid H3PO4 in a common acid bath. The produced gaseous CO2 was purified with a manual vacuum line before introduction into a Thermo Scientific MAT 253 dual-inlet mass spectrometer. Each purified CO2 gas was analyzed for its abundance in isotopologues with m∕z from 44 to 49 versus a working gas provided by Oztech Trading Corporation with ‰ Vienna Pee Dee Belemnite (VPDB) and ‰ Vienna Standard Mean Ocean Water (VSMOW), as determined with the international reference material NBS19. One single Δ47 measurement corresponds to 70 cycles of 26 s integration time each (total integration time = 1820 s). Conventional δ18O and δ13C data were also acquired simultaneously with Δ47 measurements with this instrument (Tables S1 and S2). They are in excellent consistency with data obtained with the continuous-flow method on smaller samples (Table S1).
The Δ47 is calculated as a function of the stochastic distribution of the CO2 isotopologues, as follows:
where Δ47 is expressed in per mill (‰), and R47, R46 and R45 are the abundance ratios of the masses 47, 46 and 45, respectively, relative to the mass 44 (12C16O16O). denotes measured ratios of the CO2 sample. values are calculated from the measured 44, 45, 46 and 47 abundance ratios. The number of isotopologues of mass 47 (mainly 13C18O16O, but also 12C17O16O and 13C17O17O) measured within the CO2 sample extracted from the acid digestion of the carbonates is linked to the number of isotopologues of mass 63 (mainly 13C18O16O16O) within the reacted carbonate mineral (Guo et al., 2009). For the correction from 17O interferences, we used the 17O correction parameters from Brand et al. (2010), as recently recommended (Daëron et al., 2016; Schauer et al., 2016). In order to transfer the obtained raw Δ47 data into the absolute carbon dioxide equilibrium scale “CDES” (Δ47 CDES90 being the Δ47 values of carbonates reacted within acid at 90 ∘C), standards of CO2 gases equilibrated at 25 and 1000 ∘C and with bulk isotopic compositions covering the range of measured carbonate samples (δ47 values between −50 ‰ and +24 ‰) were analyzed interspaced with unknown samples (typically 15 equilibrated CO2 gas analyses by discrete session of analysis, four analytical sessions in total; Table S5). For each analytical session, as recommended in Dennis et al. (2011), the Δ47 data were finally corrected with a fixed equilibrated gas line slope (only slightly varying from 0.0048 to 0.0062 over our analytical sessions) and an empirical transfer function (slopes varying from 1.0859 to 1.1344) based on the equilibrated CO2 standards. Finally, the accuracy of our whole dataset and processing procedure was validated on carbonate reference material (i.e., IPGP-Carrara and 102-GC-AZ01), typically analyzed every two unknown samples (Table S5). The Δ47 values obtained at IPGP over the course of this study are ‰ (1 SD, n=16) for IPGP-Carrara and ‰ (1 SD, n=18) for 102-GC-AZ01. Those values are indistinguishable from the values obtained at IPGP over 4 years of analyses on the same instrument (n>300) or previously reported by other laboratories (Daëron et al., 2016).
2.3 Temperature estimates and associated uncertainties
Apparent temperatures obtained from oxygen isotope compositions were calculated based on the measured δ18Ocarbonate values of both the precipitated carbonate and the precipitation water in each experimental vial (Table S1) and using the equation of oxygen isotopes' fractionation between calcite and water from Kim and O'Neil (1997). Apparent temperatures issued from clumped isotope compositions were calculated from Δ47 CDES90 data using the composite universal Δ47–T calibration (Eq. 3 from Bonifacie et al., 2017, with T, the temperature). It is noteworthy that our main observations and conclusions do not change if other calibrations to temperature are used for δ18O and/or Δ47 (e.g., Coplen, 2007; Kelson et al., 2017) (see also Table S3). For both proxies, the uncertainties on temperature estimates reported here correspond to the standard deviation of the mean of replicated isotopic measurements of the same powder propagated in the calibration equation (but the actual errors on the calibration themselves are not considered). Note that the long-term external reproducibility on homogeneous calcite reference materials found in this study (i.e., ±0.020 ‰, 1 SD) is used for samples with only one measurement or with 1 SD lower than 0.020 ‰ (Tables S1 and S5, Sect. S1 in the Supplement).
For the temperature (T) derived from the Δ47 data, we chose the calibration determined by Bonifacie et al. (2017) as it integrates a consequent number of data (n>300), whose statistical weight has been properly considered and covers a wide temperature range (from 1 to 350 ∘C), three characteristics that were recently shown by several teams as governing the precision on Δ47–T calibration equations (Bonifacie et al., 2017; Kelson et al., 2017; Fernandez et al., 2017). Importantly, this calibration covers the high apparent temperature ranges reported here (i.e., low Δ47 values) allowing us to avoid loss of precision and accuracy when extrapolating to temperature ranges that have not been experimentally investigated. Finally the Bonifacie et al. (2017) calibration has been checked independently with other methods (Mangenot et al., 2017; Dassié et al., 2018) on the range of temperatures (∼30 to 96 ∘C) where most available calibrations diverge and/or are not well constrained. Indeed, these studies report excellent consistencies: (i) between TΔ47 and homogenization temperatures from fluid inclusion microthermometry (Mangenot et al., 2017) and (ii) between the δ18Owater values directly measured in fluid inclusions by cavity ring-down spectroscopy and those calculated from combined TΔ47 and δ18Ocarbonate of the host mineral (Dassié et al., 2018). Though we recognize that the normalization to carbonate standards presented in Bernasconi et al. (2018) might become commonly used by the community in the future (i.e., with the ongoing intercomparison InterCarb project), we preferred not to use this correction frame here because not enough of the four carbonate standards proposed by Bernasconi et al. (2018) were run together with our samples (n=14 runs in total of ETH1, ETH2, ETH3 and ETH4 standards; Table S5), and such a normalization method will then introduce larger uncertainty than the normalization we performed with the large number of equilibrated gases ran daily together with our unknowns (n=104 equilibrated gas; Table S5 – note also 33 secondary carbonate standards 102-GC-AZ01 and IPGP-Cararra, also run in other IPGP studies and some other laboratories). Also remarkably, Δ47 values obtained here on the four ETH carbonate standards are all systematically higher than values reported in Bernasconi et al. (2018) (Table S4). Though the reason for this positive offset is still unclear, it is noteworthy that positive offsets are also observed when compiling other recent published values (Table S4; Daëron et al., 2016; Schauer et al., 2016; Fiebig et al., 2019).
3.1 Δ47 and δ18O compositions of microbial carbonates can present strongly correlated vital effects
We performed Δ47 measurements on (i) microbial carbonates precipitated without CA by faithfully replicating the experiment detailed in Thaler et al. (2017) and (ii) microbial carbonates precipitated in the presence of CA remaining from these experiments. These calcium carbonates were precipitated as the result of microbially driven hydrolysis of urea into DIC and ammonia (Millo et al., 2012). They constitute a reliable model for carbonate precipitation triggered by enzymatic production or transport of DIC, as is the case for micro- and macro-skeletal carbonates common in the Phanerozoic and for microbially mediated carbonates since the Precambrian.
Without CA, the isotopic values of the very first carbonate precipitates present strong isotopic offsets from equilibrium values. The Δ47 offset to equilibrium starts from −0.270 ‰ (the largest Δ47 offset ever measured in solid carbonates), and the offset to equilibrium reaches −24.7 ‰ for δ18Ocarbonate (Fig. 3 and Table S1). Both Δ47 and δ18Ocarbonate absolute values then increase as ureolysis progresses, reducing offsets from equilibrium values to −0.179 ‰ for Δ47 and −15.7 ‰ for δ18Ocarbonate. In the presence of CA, the trends observed for the Δ47 and δ18Ocarbonate values are similar, but the offsets from equilibrium are drastically reduced (down to −0.027 ‰ for Δ47 and −1.4 ‰ for δ18Ocarbonate at the end of the experiment; Fig. 3), hence attesting for ongoing isotopic equilibration of DIC with water by CA enzymatic activity prior to and during carbonate precipitation. The comparable behavior of Δ47 and δ18Ocarbonate values with respect to CA suggests that both disequilibria are inherited from the δ18O and Δ47 signatures of the DIC generated by the biological activity.
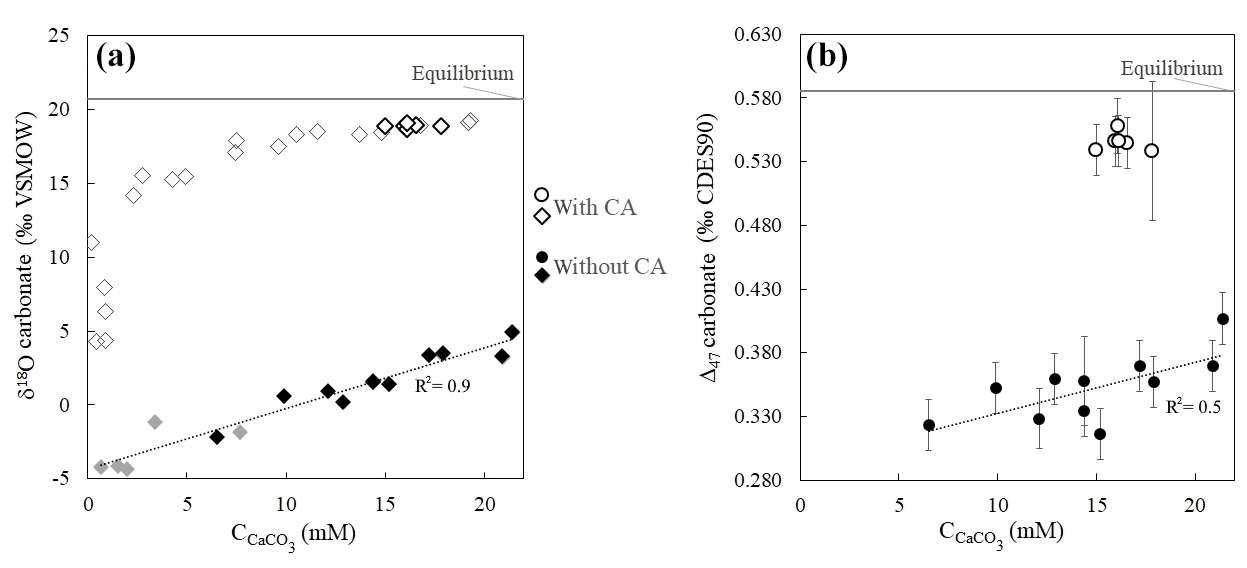
Figure 3Strong δ18O and Δ47 disequilibria recorded in microbial carbonates as shown by δ18Ocarbonate (a) and Δ47 (b) values of calcium carbonates (CaCO3) precipitated during bacterial ureolysis at 30 ∘C (with and without carbonic anhydrase, CA; open and solid symbols, respectively) as a function of carbonate accumulation (). Black symbols correspond to samples for which both Δ47 and δ18O measurements were performed. The grey horizontal lines are equilibrium δ18Ocarbonate and Δ47 values at 30 ∘C for calcite following Bonifacie et al. (2017) and Kim and O'Neil (1997) calibrations, respectively. Uncertainties (1 standard deviation, 1 SD) are smaller than the symbol for δ18O and values (Table S1). Reported Δ47 uncertainties are detailed in Sect. 2 and Sect. S1 in the Supplement.
3.2 Δ47 and δ18Ocarbonate disequilibrium originates from the metabolic production of DIC
Here, we discuss the potential processes known to generate δ18O and Δ47 isotope fractionations during carbonate precipitation and we identify the main mechanism explaining our paired Δ47 and δ18Ocarbonate disequilibria. The relatively high precipitation rate (R) in our experiments ( mol m−2 s−1; Thaler et al., 2017) can only account for an oxygen kinetic isotope fractionation (KIF) of about 1 ‰ to 2 ‰ for δ18O values (Watkins et al., 2013), while the oxygen isotope disequilibrium recorded in our carbonates reaches −24.7 ‰. Degassing of CO2, known to fractionate DIC oxygen isotopes (Affek and Zaarur, 2014), can be ruled out as there is no gas phase in our experiments (see Sect. 2.1). Any potential kinetic fractionation due to DIC diffusion (Thiagarajan et al., 2011) is also unlikely as precipitation occurred on the bacterial DIC-producing cells, as highlighted by scanning electron microscopy showing bacterial cells trapped within and at the surface of carbonate crystals (Fig. 1). Accordingly, the large offsets from equilibrium values observed for both Δ47 and δ18O in our microbial carbonates can only result from (i) a KIF induced by CO2 hydration or hydroxylation into (but only if ureolysis produces CO2 rather than H2CO3, which has not been established yet; Matsuzaki et al., 2013) or (ii) a metabolic isotopic signature of the DIC produced by the bacteria, inherited from the initial isotopic composition of urea and/or due to a KIF introduced by the urease enzyme. CO2 hydration and hydroxylation lead to the formation of , with two oxygen atoms coming from CO2 and the third one from H2O (hydration) or OH− (hydroxylation). The δ18O value can then be estimated using a simple mass balance calculation (Létolle et al., 1990; Usdowski et al., 1991). The newly formed is depleted in 18O compared to the reacting CO2 because of the incorporation of oxygen coming from H2O or OH−, both depleted in 18O relative to 16O in contrast to CO2 (Green and Taube, 1963; Beck et al., 2005). Such a low δ18O value, several per mill lower than the equilibrium one, can then be preserved in the calcium carbonate if precipitation occurs shortly after CO2 hydration and hydroxylation and before the full equilibration with water (Rollion-Bard et al., 2003). Regarding clumped isotopes, ab initio calculations predict that the fractionation associated with CO2 hydration and hydroxylation increases the relative abundance of 13C–18O bonds and thus the Δ47 value (Guo, 2009). Even though this predicted fractionation trend has previously been used to explain several datasets for which CO2 hydroxylation was assumed to occur prior to carbonate precipitation (Tripati et al., 2015; Spooner et al., 2016), such a tendency can only be validated using data acquired on carbonates for which CO2 hydration and hydroxylation are demonstrated. This is the case of (i) hyperalkaline travertines (Falk et al., 2016) even though part of the reported kinetic isotope fractionation can be interpreted as resulting from the CO2 dissolution process (Clark et al., 1992) and (ii) two experimental samples (Tang et al., 2014) precipitated at high pH where CO2 hydroxylation dominates. Both studies show, in agreement with the ab initio calculations (Guo, 2009), higher Δ47 and lower δ18Ocarbonate values compared to equilibrium. Thus, in a case where ureolysis would produce CO2 in isotopic equilibrium with water, the Δ47 values affected by CO2 hydration and hydroxylation recorded in calcium carbonates should be higher than the equilibrium value, while our microbial carbonates show Δ47 values lower than equilibrium. Thus, we conclude that our low Δ47 values measured in carbonates can only be explained by a metabolic source effect. In our case it corresponds to the ureolytic production of DIC, either directly as H2CO3 or as CO2, with a Δ47 value low enough to compensate for any potentially succeeding increase due to the KIF associated with CO2 hydration and hydroxylation. Nonetheless, the slow but continuous increase observed in our experiment without CA for both Δ47 and δ18Ocarbonate values more likely reflects ongoing equilibration of DIC oxygen isotopes with water at a slow rate.
Our results highlight that the isotope clumping proceeds continuously as C–O bonds are breaking and reforming in the DIC, allowing oxygen isotopes (16O, 17O and 18O) to be redistributed between H2O, OH−, H2CO3, and species via attachment to CO2 and detachment from . In the experiment with CA, both Δ47 and δ18Ocarbonate simultaneously reach values close to equilibrium, and without CA both Δ47 and δ18Ocarbonate values increase simultaneously. This coevolution corroborates former observations of comparable kinetics for clumped isotopes and δ18O equilibration between DIC and water or CO2 and water once δ13C is equilibrated (Affek, 2013; Clog et al., 2015). This principle has been used to correct for the disequilibrium fractionation factor in speleothems (Affek et al., 2008).
3.3 Erroneous yet comparable temperatures reconstructed from disequilibrium Δ47 and δ18O values in carbonates
Apparent temperatures were calculated from disequilibrium Δ47 values obtained in the experiment without CA using the calibration of Bonifacie et al. (2017). Ranging from 198±21 to 115±8 ∘C (Fig. 4), they are at odds with the actual precipitation temperature of 30±1 ∘C (see Sect. 2.1). This shows that when carbonates precipitate from DIC in oxygen isotope disequilibrium with water, the abundance of 13C–18O bonds in carbonates does not correlate with precipitation water temperature. Conversely, the temperatures reconstructed from the Δ47 values of carbonates precipitated in the presence of CA, ranging from 47±6 to 39±2 ∘C, are much closer to the actual precipitation temperature. Interestingly, the apparent temperatures reconstructed using Kim and O'Neil (1997) calibration from the δ18Ocarbonate and δ18Owater values of the same samples show comparable offsets from the actual temperature in both experiments without CA (from 218±2 to 139±1 ∘C) and with CA (from 39±1 to 37±1 ∘C) (Fig. 4). Practically, this implies that similar temperatures calculated from both carbonate Δ47 and δ18Ocarbonate values (in a case where the precipitation water δ18O can be determined) can neither constitute evidence against O isotope disequilibrium nor confirm that this is the true precipitation temperature.
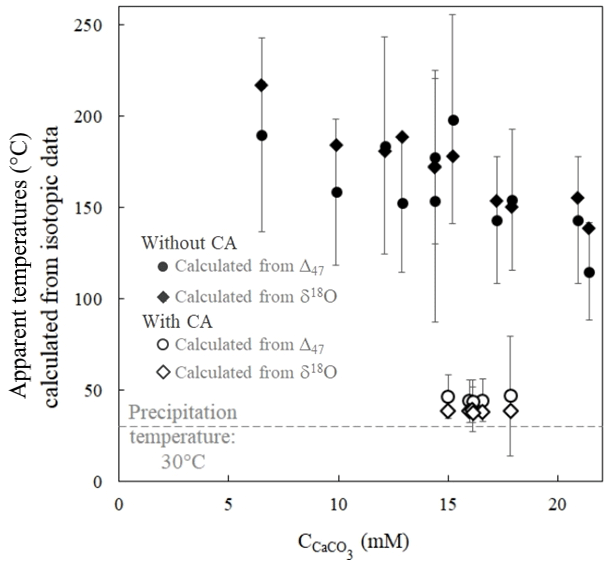
Figure 4δ18Ocarbonate and Δ47 disequilibria in microbial carbonates induce comparable biased estimates of precipitation temperature as illustrated by apparent temperatures calculated from the carbonate δ18Ocarbonate and Δ47 signatures as a function of CaCO3 accumulation. Open and solid symbols refer to the experiments with and without CA, respectively. The dashed grey line corresponds to the actual precipitation temperature. Apparent temperatures are respectively calculated from the δ18Ocarbonate and Δ47 calibrations to the temperature of Kim and O'Neil (1997) and Bonifacie et al. (2017). Reported uncertainties were calculated as the propagation of the 1 standard deviation (1 SD) error of the isotopic data in the calibration equations (Sect. 2 and Sect. S1 in the Supplement).
3.4 Δ47 and δ18Ocarbonate paired disequilibria record the δ18O of the water in which the carbonates precipitated
The fact that both Δ47 and δ18Ocarbonate values permit us to calculate similarly evolving apparent temperatures along the (dis)equilibration profile recorded in carbonates as the experiment proceeds indicates that the δ18Ocarbonate, δ18Owater, Δ47 and apparent temperature values are all together linked. In a diagram of Δ47 versus δ18Ocarbonate, all of our data align, irrespectively of the fact that they are in strong isotopic disequilibrium or close to equilibrium (Fig. 5). Their alignment is fitted with what would be expected for equilibrium Δ47 and δ18Ocarbonate values of calcite precipitated at various temperatures from water at a given δ18Owater value. This δ18Owater value can be calculated by combining, for the same temperature, the equations of Δ47 and δ18Ocarbonate temperature calibrations from Bonifacie et al. (2017) and Kim and O'Neil (1997), respectively (Eq. 2):
with δ18Owater and δ18Ocarbonate values in the same isotopic reference (here VSMOW) and Δ47 values reported on the absolute carbon dioxide equilibrated scale (Δ47 CDES90). The calibration of Kim and O'Neil (1997) was preferred over more recent calibration equations (e.g., Watkins et al., 2013) because it provides the best consistency for temperatures reconstructed from both the carbonate δ18O and Δ47 values at temperatures above 100 ∘C. Note that Kim and O'Neil (1997) and Bonifacie et al. (2017) calibrations were developed independently, which prevents circular reasoning. Finally, as the calibration of Kim and O'Neil (1997) is the most used calcite calibration to date, it also allows for a broader comparison with previously published results.
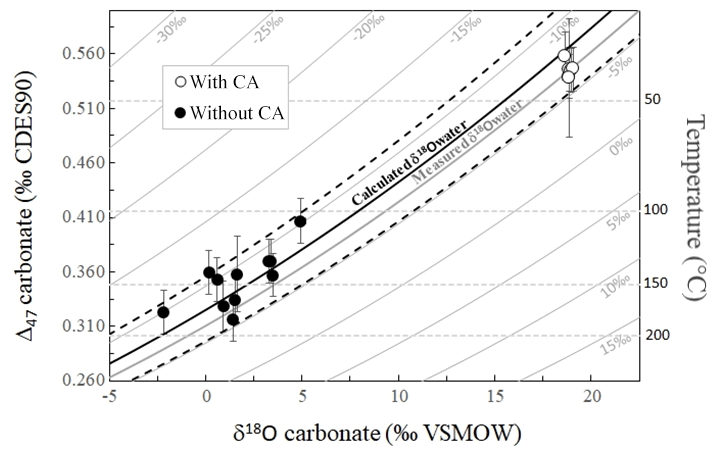
Figure 5Combined δ18Ocarbonate and Δ47 disequilibria of microbial carbonates precipitated at 30 ∘C allow reconstruction of the δ18O of the water (δ18Owater) in which they precipitate. Solid grey curves represent the calculated Δ47 and δ18Ocarbonate compositions of carbonates precipitated at oxygen isotope equilibrium from water with fixed δ18Owater values (indicated on each curve) and variable temperatures. Horizontal dashed grey lines are calculated for fixed temperatures and variable δ18Owater. The average δ18Owater value of ‰ measured in our experiments is reported using the thick solid grey curve. The solid black curve was obtained using the δ18Owater calculated with Eq. (2) ( ‰) with its associated errors (dashed black curves).
Despite the fact that the data present a large range of offsets from equilibrium (Fig. 5), the mean δ18Owater value calculated using Eq. (2) for each combination of Δ47 and δ18Ocarbonate values measured for our carbonates is ‰ (1 SD), indistinguishable (i.e., within errors) from the δ18Owater values measured in our experiments ( ‰ with CA and ‰ without CA; Fig. 5). Note that such precision in δ18Owater values found in disequilibrium carbonates is remarkable considering that even for equilibrium carbonates at isotopic equilibrium for both δ13C and δ18O, δ18Owater can only be retrieved from paired Δ47 and δ18Ocarbonate values with a precision of ±1 ‰ at best (see Fig. S1 in the Supplement). This creates the promising opportunity to retrieve the δ18O value of the water in which carbonates precipitated out of equilibrium for both δ18Ocarbonate and Δ47.
In order to evaluate the applicability of such an approach to other types of carbonates, Fig. 6 compiles disequilibrium paired δ18Ocarbonate and Δ47 data from two previously published experimental studies (Tang et al., 2014; Staudigel and Swart, 2018). These studies were chosen to further evaluate the relevancy of our δ18Ocarbonate–Δ47 correlation because they are the only published dataset reporting full sets of measured (rather than calculated) δ18Owater, δ18Ocarbonate and Δ47 values, with one or both proxies showing disequilibrium, together with precipitation temperatures. A perfect knowledge (i.e., measurements and not estimates) of these four parameters is mandatory here to adequately test whether the use of our new δ18Owater proxy could be generalized to a large diversity of carbonates. This thus precludes plotting most published Δ47 studies on both natural and experimental samples, in which δ18Owater and/or temperature were not directly measured, in Fig. 6. These two datasets are also recent enough to allow the conversion of their Δ47 values to the currently used normalization method (i.e., the CDES absolute reference frame). It will then allow comparison with future studies, if measuring and reporting all these four parameters together becomes the rule rather than the exception in Δ47 studies.
Figure 6a shows paired δ18Ocarbonate and Δ47 values of abiotic carbonates produced at 5, 25 and 40 ∘C that are known to be affected by KIF due to fast precipitation and for at least two of them by KIF due to CO2 hydration and hydroxylation prior to precipitation (Tang et al., 2014). Except for these two carbonate samples, the data align on a covariation curve of Δ47 versus δ18Ocarbonate that cannot be explained solely by temperature variation. As for our microbial carbonates obtained with or without CA, the average calculated δ18Owater (Eq. 2; ‰) matches within error with the measured δ18Owater ( ‰) (Dietzel et al., 2009).
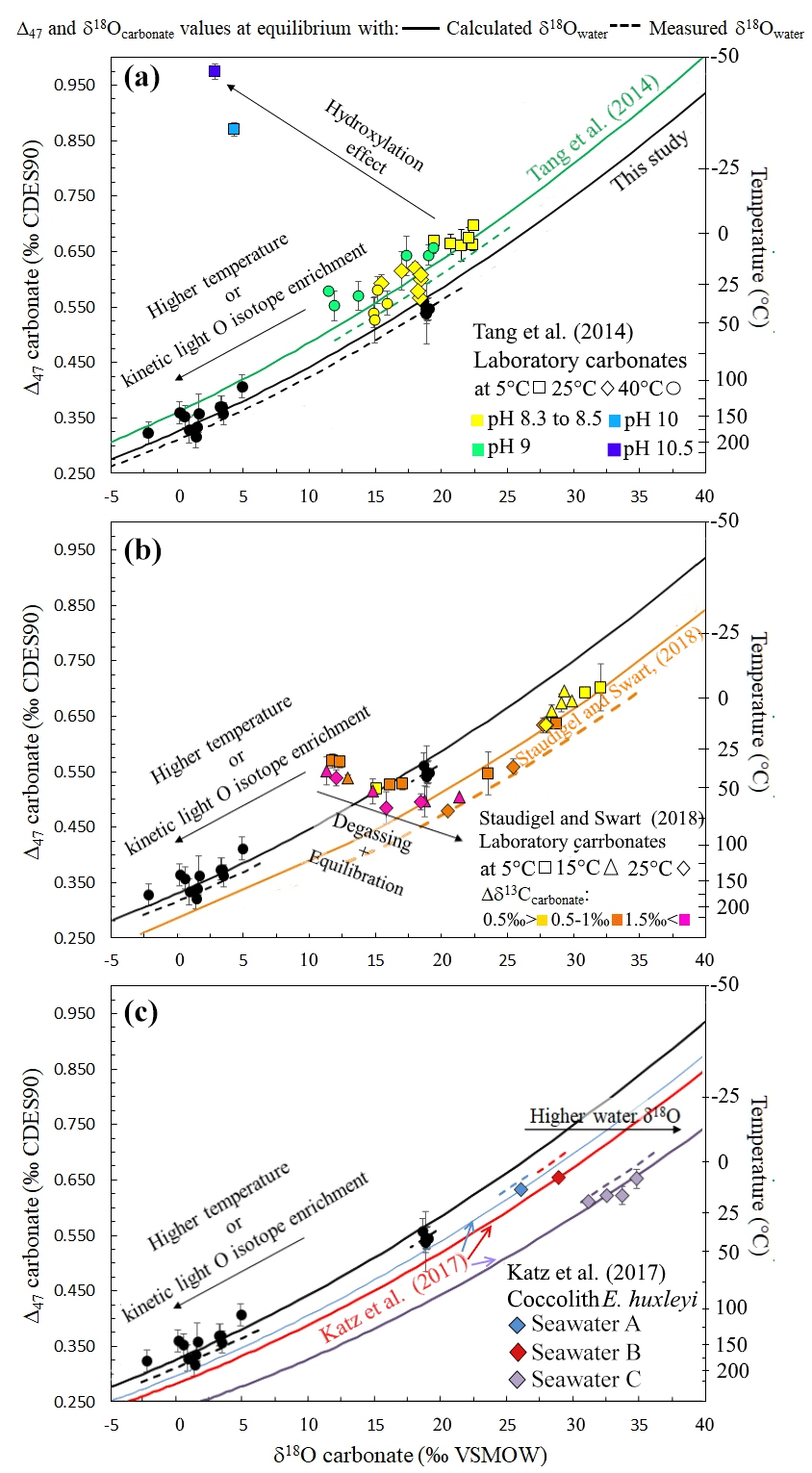
Figure 6Δ47 and δ18Ocarbonate relationship to precipitation water δ18Owater for other solid carbonates presenting oxygen isotope disequilibria. In (a) to (c) the black data series (this study, performed at 30 ∘C) shows how kinetic oxygen isotope fractionation in the DIC prior to carbonate precipitation can be mistaken for high-temperature isotopic equilibrium. Similarly to Fig. 5, the solid curves were obtained using the δ18Owater calculated with Eq. (2). (a) Abiotic carbonates from Tang et al. (2014) illustrating the effect of CO2 hydroxylation on Δ47 and δ18Ocarbonate values (various pH values plotted with different colors, various temperatures plotted with different symbols). (b) Abiotic carbonates from Staudigel and Swart (2018) illustrating the effect of CO2 degassing and DIC oxygen isotope equilibration with water on Δ47 and δ18Ocarbonate values. Δδ13Ccarbonate stands for the difference between the δ13C value measured in carbonates and the final δ13C of the data series at the end of equilibration (various Δδ13C ranges plotted with different colors, various temperatures plotted with different symbols). (c) Coccolithophorid E. huxleyi grown at 7, 10, 15, 20 and 25 ∘C from Katz et al. (2017) showing how coccoliths with equilibrium Δ47 values record the equilibrium δ18Owater of their body water, which differs from that of the culture medium (i.e., artificial seawaters A, B and C plotted with different colors).
Figure 6b shows paired δ18Ocarbonate and Δ47 values of abiotic carbonates that were precipitated during an initial CO2 degassing + DIC equilibration phase followed by solely equilibration with water at 5, 15 and 25 ∘C (Staudigel and Swart, 2018). During the latter equilibration phase, even though the carbonates precipitated out of isotopic equilibrium, the paired δ18Ocarbonate and Δ47 values align on a covariation curve of the average calculated δ18Owater value (Eq. 2; ‰) close to the measured δ18Owater (−0.65 ‰). As a major outcome of this study, we thus anticipate that reliable δ18Owater values of precipitation water can be retrieved from carbonates presenting Δ47 and δ18Ocarbonate values in strong disequilibrium.
Some data presented in Fig. 6 also permit us to evaluate the conditions of applicability of our approach. In Fig. 6a, the two data points deviating from the covariation curve of Δ47 versus δ18Ocarbonate correspond to carbonates precipitated at pH ∼10 and 5 ∘C (while the others formed at pH and temperatures ranging from 8.3 to 9 and 5 to 40 ∘C, respectively) that have recorded a KIF due to CO2 hydration and hydroxylation prior to precipitation (Tang et al., 2014). At pH = 10, CO2 reacts at 95 % with OH−, and at 5 ∘C DIC isotopic equilibration with water takes days. To a lesser extent, the KIF induced by CO2 hydroxylation also seems visible at pH = 9 (and 40 ∘C), where CO2 reacts at 82 % with OH−, but DIC isotopic equilibration with water at 40 ∘C only takes about 15 h. As previously detailed, the direction of these isotopic offsets from equilibrium is compatible with ab initio calculations (Guo, 2009) and can be intuitively understood as follows: in carbonates derived from CO2 hydroxylation, the term used for the Δ47 calculation (Eq. 1) should be strongly modified as the 18O concentration in OH− and H2O is lower than in CO2, and the reaction does not add more 13C than what is present in CO2. This might explain why in the case of disequilibria acquired through CO2 hydroxylation, the correlation between paired δ18O and Δ47 disequilibria and the precipitation water δ18O is not preserved, and δ18Owater cannot be reconstructed by the approach proposed here. The negative slope associated with this KIF on the Δ47 and δ18Ocarbonate diagram (Fig. 6a) is nevertheless a good tool to identify CO2 hydroxylation reactions.
In Fig. 6b, during the CO2 degassing phase of the precipitation experiment (Staudigel and Swart, 2018), the data also deviate from the covariation curve of δ18O versus Δ47. This behavior was interpreted by the authors as a decoupling between Δ47 and δ18Ocarbonate values due to variable kinetics of 12C–O and 13C–O bonding. A known difference in equilibration kinetics takes place between C and O isotopes in the carbonate system as carbon isotopes equilibrate in seconds, while oxygen isotopes necessitate minutes to hours to equilibrate among the different oxygen-bearing species (i.e., CO2, , , H2O, OH−), depending on the pH, temperature and salinity of the solution (Zeebe and Wolf-Gladrow, 2001). However, note that in that experiment, the carbon isotope compositions evolved for several hours as a result of CO2 degassing (Staudigel and Swart, 2018). We propose here that CO2 degassing, because it affects both C and O isotopes, modifies the term (in Eq. 1), thus preventing Δ47 and δ18O from varying with the proportionality that allows us to retrieve the δ18Owater value on a covariation plot of Δ47 versus δ18O. Hence, as for CO2 hydroxylation, in the case of a KIF induced by CO2 degassing, δ18Owater cannot be reconstructed exclusively from disequilibrium δ18Ocarbonate and Δ47 values.
In summary, we conclude that mechanisms that can drastically change the term in Δ47 calculation (such as CO2 hydroxylation and degassing) prevent δ18Owater reconstructions from paired disequilibrium Δ47 and δ18Ocarbonate values. Nevertheless, these mechanisms lead to peculiar types of carbonates (e.g., speleothems that form in caves from CO2 degassing, and travertine that forms on lands where fluids and gas escape from subsurface reservoirs, for CO2 hydroxylation) that represent only a small fraction of all the carbonates existing on Earth. We hypothesize that ureolysis, which consists in two successive steps of urea hydrolysis, an exchange reaction with the H2O molecule from the aqueous medium, might give a DIC whose term in the Δ47 calculation is already close to that of DIC under equilibration with the δ18Owater. This would explain why even our most extreme out-of-equilibrium carbonates still fall close to the covariation line of Δ47 versus δ18Ocarbonate corresponding to the real δ18Owater value.
3.5 Toward a better understanding of body water δ18O in biomineralizing organisms
The ability to reconstruct precipitation water δ18Owater from disequilibrium Δ47 and δ18Ocarbonate values further allows us to examine the origin of the vital effect observed in organisms for which (i) CO2 degassing and hydration and hydroxylation KIF can be ruled out, and (ii) only small δ13C variations are observed, thus preserving the term in Δ47 calculation. We hypothesize that such an approach could help to understand how Δ47 and δ18O signals are affected by kinetic effects in most of the biogenic carbonates, provided that CO2 hydroxylation or degassing does not occur prior to carbonate precipitation. This approach could thus be applied to the vast majority of sedimentary carbonates (Milliman, 1993) (i.e., microbialites, brachiopods, bryozoans, bivalves, foraminifera, coccoliths) and since deep time, even when δ18Ocarbonate variations occur in the shell of the organism. Additionally, the data presented here stand as an experimental demonstration that the mechanisms controlling carbonate δ18O equilibration with water (i.e., DIC equilibration with water) also control solid carbonate Δ47 equilibrium (Watkins and Hunt, 2015). This result can be used to recover information on biomineralization mechanisms. For example, in recent coccolithophorid Emiliania huxleyi culture experiments, the calcitic shell produced by the organism systematically yields a 2 ‰ positive δ18O offset from equilibrium values while its Δ47 values seem to faithfully record precipitation temperature (Katz et al., 2017). These coccolithophorids were grown in waters with different δ18Owater compositions (i.e., measured at −6.14 ‰, −5.82 ‰ and 0.65 ‰ VSMOW that are respectively seawater A, B and C in Fig. 6c). Based on our results, which demonstrate that no δ18O disequilibrium should be recorded in solid carbonates if the associated Δ47 is at equilibrium, we can assume that the coccoliths precipitated at oxygen isotope equilibrium. We also calculate the actual δ18O value of the water in which precipitation took place from Eq. (2) (respectively shifted by 1.0±0.2 ‰, 2.1±0.4 ‰ and 1.1±0.7 ‰ towards more positive values compared to the δ18Owater value measured for the culture medium water; Fig. 6c). This could reflect a biologically driven difference between the δ18O of body water at the precipitation site inside E. huxleyi and the δ18O of ambient water (i.e., the culture medium water). This hypothesis is supported by what is known about intracellular precipitation of coccoliths performed by coccolithophorids: each coccolith forms from the accumulation of coccolithosomes, which are vesicles containing up to approximately twelve 7 nm spherical calcium-rich granular units (Outka and Williams, 1971). Water in these ∼100 nm vesicles can be considered a finite reservoir whose isotopic composition could be modified through isotopic exchange with DIC affected by metabolic isotope fractionation. Another mechanism that could increase the δ18O value of a finite water reservoir by equilibrating it with a comparable reservoir of DIC would be the introduction of DIC systematically as CO2. As and are enriched in 16O in comparison to CO2, the CO2 conversion to and at equilibrium before precipitation would pump 16O from water.
In both case scenarios, a local change in water isotopic composition requires that the water molecules' turnover (i.e., external inputs) in these cellular organites is slow enough. Coccolithosomes are subunits of the Golgi complex, which is a system of flat stacked vesicles concentrating a lot of membranes in a small location (Outka and Williams, 1971). It is thus plausible that in a single-celled organism, living in seawater and performing intracellular biomineralization, specific osmolarity and water circulation regulation mechanisms are occurring. It is particularly plausible in the Golgi complex, whose water content is isolated from seawater by several membranes. We thus suggest that inside coccolithosomes, coccolith precursors precipitate in equilibrium with the body water for oxygen isotopes but that the body water has a different δ18O value than the seawater, which explains the observed δ18O apparent fractionation while the Δ47 composition reflects culture temperature (Katz et al., 2017). It has already been highlighted through geochemical analysis of coccoliths that coccolithosome water has altered pH (Liu et al., 2018) and ion concentrations (Hermoso et al., 2017) in comparison to seawater. We hypothesize that the internal δ18O water would thus be another parameter controlled by the coccolithophore algae.
3.6 Ubiquity of the observed δ18Ocarbonate–δ18Owater–Δ47–temperature covariations in both equilibrium and disequilibrium carbonates
As shown above (Fig. 5), in a diagram of Δ47 versus δ18Ocarbonate, disequilibrium carbonates precipitated at fixed temperature plot on the theoretical line of equilibrium carbonates precipitated with a similar δ18Owater but at a different temperature. This is illustrated in Fig. 7 where the three disequilibrium data series studied in this paper (Fig. 5 for this study and Fig. 6a and b for datasets from Tang et al., 2014, and Staudigel and Swart, 2018) align with equilibrium data series. In other words, the values of the disequilibrium 1000ln αcarbonate−water for oxygen isotopes (with ) are similar to the equilibrium 1000ln αcarbonate-water for any given, and independently determined, apparent Δ47 temperature (Fig. 7). In detail, our closest-to-equilibrium data recording low apparent temperatures match better with the predicted equations from Coplen (2007) and Watkins et al. (2013), recently updated (Daëron et al., 2019). This last calibration is based on carbonates from two caves where calcite precipitates extremely slowly and is thus assumed to have precipitated at equilibrium. Note that the use of these two cave samples for determining the dependence on temperature of the equilibrium 1000ln αcarbonate-water relies on the assumption that constant environmental conditions, including temperature in the two caves (7.9 and 33.7 ∘C) and the δ18Owater value of the precipitation water, prevailed over the whole period of carbonate precipitation (Coplen, 2007; Kluge et al., 2014). In Fig. 7, the disequilibrium data recording high apparent temperatures (above 100 ∘C) match better with the predicted equation of Kim and O'Neil (1997). This 1000ln αcarbonate-water dependence on temperature was established on carbonates precipitated in the laboratory at well-known δ18Owater and temperatures (from 10 to 40 ∘C) but was suspected to present a small KIF due to a high precipitation rate that lowers the value of the 1000ln αcarbonate-water (Watkins et al., 2013). Despite this, we used this equation to retrieve the δ18Owater from our experimental carbonates, because most of them are associated with high apparent Δ47 temperatures. Coplen (2007) or Watkins et al. (2013) equations would have 2 ‰ lower values (ca. ‰ compared to ‰ calculated with the equation of Kim and O'Neil, 1997). From our results, due to our experimental conditions and the associated error in our dataset, it is not possible and not our intention to argue in favor of one of these calibrations. This however shows how crucial it is to improve knowledge on the equilibrium 1000ln αcarbonate-water at both high and low temperatures in order to improve the accuracy and precision of our new proxy for reconstructing the δ18Owater from which carbonates, even disequilibrium ones, precipitated.

Figure 7The relation to temperature of the equilibrium oxygen isotope fractionation factor between calcium carbonate and water (1000ln αcalcite−water) appears to be retrievable from solid carbonates (mainly calcites) in strong clumped and oxygen isotope disequilibrium such as our microbial carbonates (black dots, precipitated at 30 ∘C) and two additional data series of laboratory-grown carbonates showing disequilibrium fractionation (Tang et al., 2014; Staudigel and Swart, 2018) (green and yellow dots, respectively). The data points affected by CO2 hydroxylation (Tang et al., 2014) or CO2 degassing (Staudigel and Swart, 2018) (see Fig. 6) are not included. Grey symbols correspond to cave carbonates precipitated at or near equilibrium (Coplen, 2007; Daëron et al., 2019) or laboratory experiments (Kim and O'Neil, 1997; Watkins et al., 2013). Those grey data series are usually considered representative of the equilibrium fractionation factor between calcium carbonate and water whose relations to temperature, extrapolated at high temperature, are illustrated by the different dashed curves. Plotted temperatures correspond to precipitation temperatures except for disequilibrium carbonates for which apparent temperatures have been calculated based on Δ47 values. The x-axis errors for this study are included in the symbol size. The y-axis error for all the reconstructed temperatures is given on the figure.
Importantly, we here establish a new method to determine the equilibrium 1000ln αcarbonate-water, which consists in using the kinetics of Δ47 and δ18O covariations during (dis)equilibration. Notably, because of the very large range of apparent temperatures recorded by disequilibrium carbonates (between ∼40 and 200 ∘C; Fig. 7), this method could be particularly adapted to calibrate 1000ln αcarbonate-water at high temperatures for which the differences between the two most popular equations for 1000ln αcarbonate-water dependence on temperature (Kim and O'Neil, 1997; Coplen, 2007) appear larger (Fig. 7). Unfortunately, none of the three experimental setups having produced these disequilibrium carbonates (this study, as well as Tang et al., 2014, and Staudigel and Swart, 2018) were designed for the purpose of calibrating the equilibrium 1000ln αcarbonate-water. It is thus not possible, using these datasets, to propose a meaningful calibration. At least in our experiment, too many phenomena including the relatively high precipitation rate, variations in δ13C values (∼3 ‰) (Thaler et al., 2017) and presence of traces of aragonite and vaterite in our carbonates (Sect. 2.1 and Sect. S1 in the Supplement) lower the accuracy of the reconstructed equilibrium 1000ln αcarbonate-water values.
From a broader perspective, we anticipate that such an approach will help in determining critical equilibrium fractionation factors for other gaseous isotopic systems (such as isotopologues of molecules containing S–O bonds) or minerals of prime interest in biology and geology if clumped isotope measurements expand further beyond gaseous mass spectrometry (e.g., bonding between Fe–O, Fe–S, Ca–C).
Our experimental results show that the information held in disequilibrium (and apparent disequilibrium) carbonates is diverse and promising. First, a paired Δ47 and δ18Ocarbonate disequilibrium indicates that carbonates have precipitated in a dynamic environment where DIC and water did not reach isotopic equilibrium. In our microbial carbonate experiments, all the DIC is produced in isotopic disequilibrium with water and precipitates rapidly. Accordingly, the disequilibrium O isotope compositions recorded in those carbonates are maximized compared to what can be expected in nature where newly produced DIC is expected to be mixed with at least partly equilibrated ambient DIC before carbonates precipitate. Second, the combined use of clumped and traditional oxygen isotopic compositions allows the retrieval of the δ18O of the precipitation water, i.e., organism body water or environmental water, even for carbonates presenting δ18O and/or Δ47 disequilibria or apparent disequilibria. Hence, except in the case of processes such as CO2 degassing and CO2 hydration and hydroxylation, which likely modify the term in Δ47 calculation, paired Δ47 and δ18Ocarbonates disequilibria in carbonates can be used to reconstruct the oxygen isotope composition of both DIC and water at the precipitation loci even when precipitation occurred under disequilibrium conditions. Third, the (dis)equilibration trend in a covariation diagram of Δ47 versus δ18Ocarbonates can be used as a new method to determine the equilibrium fractionation factor between carbonate and water for a wide range of temperatures. Altogether, this opens up new avenues to better constrain not only past climate changes through improved paleoenvironmental reconstructions but also the physiology and habitat of sea life sensitive to ocean acidification.
All the data generated and analyzed in this study are available within the paper and in its Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/bg-17-1731-2020-supplement.
CT and AK conceived the research. CT performed the microbial precipitation experiment and the δ13C and δ18O analyses during her PhD thesis under the supervision of MA and BM. AK performed the Δ47 analyses during her PhD thesis under MB's supervision. CT took the lead in the interpretation of the results and the writing of the original draft. All authors provided critical feedback and helped shape the research, analyses and manuscript.
The authors declare that they have no conflict of interest.
This research was supported by French MRT PhD fellowships to Caroline Thaler and Amandine Katz, the Centre de Recherches sur le Stockage Géologique du CO2 (IPGP-TOTAL-Schlumberger-ADEME) (Bénédicte Ménez and Magali Ader), and an Emergence program grant from the Paris council to Magali Bonifacie. The authors are grateful for the insightful reviews of William F. Defliese and Stefano M. Bernasconi that improved the manuscript. This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001.
This research has been supported by French MRT PhD fellowships to Caroline Thaler and Amandine Katz, the Centre de Recherches sur le Stockage Géologique du CO2 (IPGP-TOTAL-Schlumberger-ADEME) (Bénédicte Ménez and Magali Ader), and an Emergence program grant from the Paris council to Magali Bonifacie.
This paper was edited by Carolin Löscher and reviewed by William Defliese and Stefano Bernasconi.
Affek, H. P., Bar-Matthews, M., Ayalon, A., Matthews, A., and Eiler, J. M.: Glacial/interglacial temperature variations in Soreq cave speleothems as recorded by “clumped isotope” thermometry, Geochim. Cosmochim. Ac., 72, 5351–5360, https://doi.org/10.1016/j.gca.2008.06.031, 2008.
Affek, H. P.: Clumped isotopic equilibrium and the rate of isotope exchange between CO2 and water, Am. J. Sci., 313, 309–325, https://doi.org/10.2475/04.2013.02, 2013.
Affek, H. P. and Zaarur, S.: Kinetic isotope effect in CO2 degassing: Insight from clumped and oxygen isotopes in laboratory precipitation experiments, Geochim. Cosmochim. Ac., 143, 319–330, https://doi.org/10.1016/j.gca.2014.08.005, 2014.
Affek, H. P., Matthews, A., Ayalon, A., Bar-Matthews, M., Burstyn, Y., Zaarur, S., and Zilberman, T.: Accounting for kinetic isotope effects in Soreq Cave (Israel) speleothems, Geochim. Cosmochim. Ac., 143, 303–318, https://doi.org/10.1016/j.gca.2014.08.008, 2014.
Bajnai, D., Fiebig, J., Tomašových, A., Garcia, S. M., Rollion-Bard, C., Raddatz, J., Löffler, N., Primo-Ramos, C., and Brand, U.: Assessing kinetic fractionation in brachiopod calcite using clumped isotopes, Sci. Rep.-UK, 8, 533, https://doi.org/10.1038/s41598-017-17353-7, 2018.
Beck, W. C., Grossman, E. L., and Morse, J. W.: Experimental studies of oxygen isotope fractionation in the carbonic acid system at 15, 25, and 40 ∘C, Geochim. Cosmochim. Ac., 69, 3493–3503, https://doi.org/10.1016/j.gca.2005.02.003, 2005.
Bernasconi, S. M., Müller, I. A., Bergmann, K. D., Breitenbach, S. F., Fernandez, A., Hodell, D. A., Jaggi, M., Meckler, A. N., Millan, I., and Ziegler, M.: Reducing uncertainties in carbonate clumped isotope analysis through consistent carbonate-based standardization, Geochem. Geophy. Geosy., 19, 2895–2914, https://doi.org/10.1029/2017GC007385, 2018.
Bonifacie, M., Calmels, D., Eiler, J. M., Horita, J., Chaduteau, C., Vasconcelos, C., Agrinier, P., Katz, A., Passey, B. H., Ferry, J. M., and Bourrand, J. J.: Calibration of the dolomite clumped isotope thermometer from 25 to 350 ∘C, and implications for a universal calibration for all (Ca, Mg, Fe)CO3 carbonates, Geochim. Cosmochim. Ac., 200, 255–279, https://doi.org/10.1016/j.gca.2016.11.028, 2017.
Brand, W. A., Assonov, S. S., and Coplen, T. B.: Correction for the 17O interference in δ13C measurements when analyzing CO2 with stable isotope mass spectrometry (IUPAC Technical Report), Pure Appl. Chem., 82, 1719–1733, https://doi.org/10.1351/PAC-REP-09-01-05, 2010.
Burgener, L. K., Huntington, K. W., Sletten, R., Watkins, J. M., Quade, J., and Hallet, B.: Clumped isotope constraints on equilibrium carbonate formation and kinetic isotope effects in freezing soils, Geochim. Cosmochim. Ac., 235, 402–430, https://doi.org/10.1016/j.gca.2018.06.006, 2018.
Clark, I. D., Fontes, J. C., and Fritz, P.: Stable isotope disequilibria in travertine from high pH waters: laboratory investigations and field observations from Oman, Geochim. Cosmochim. Ac., 56, 2041–2050, https://doi.org/10.1016/0016-7037(92)90328-G, 1992.
Clog, M., Stolper, D., and Eiler, J. M.: Kinetics of CO2(g)–H2O(1) isotopic exchange, including mass 47 isotopologues, Chem. Geol., 395, 1–10, https://doi.org/10.1016/j.chemgeo.2014.11.023, 2015.
Coplen, T. B.: Calibration of the calcite–water oxygen-isotope geothermometer at Devils Hole, Nevada, a natural laboratory, Geochim. Cosmochim. Ac., 71, 3948–3957, https://doi.org/10.1016/j.gca.2007.05.028, 2007.
Daëron, M., Blamart, D., Peral, M., and Affek, H. P.: Absolute isotopic abundance ratios and the accuracy of Δ47 measurements, Chem. Geol., 442, 83–96, https://doi.org/10.1016/j.chemgeo.2016.08.014, 2016.
Daëron, M., Drysdale, R. N., Peral, M., Huyghe, D., Blamart, D., Coplen, T. B., Lartaud, F., and Zanchetta, G.: Most Earth-surface calcites precipitate out of isotopic equilibrium, Nat. Commun., 10, 429, https://doi.org/10.1038/s41467-019-08336-5, 2019.
Dassié, E., Genty, D., Noret, A., Mangenot, X., Massault, M., Lebas, N., Duhamel, M., Bonifacie, M., Gasparrini, M., Minster, B., and Michelot, J. L.: A newly designed analytical line to examine the reproducibility of fluid inclusion isotopic compositions in small carbonate samples, Geochem. Geophy. Geosy., 19, 1107–1122, https://doi.org/10.1002/2017GC007289, 2018.
Dennis, K. J., Affek, H. P., and Schrag, D.: Defining an absolute reference frame for “clumped” isotope studies of CO2, Geochim. Cosmochim. Ac., 75, 7117–7131, https://doi.org/10.1016/j.gca.2011.09.025, 2011.
Dietzel, M., Tang, J., Leis, A., and Köhler, S. J.: Oxygen isotopic fractionation during inorganic calcite precipitation – Effects of temperature, precipitation rate and pH, Chem. Geol., 268, 107–115, https://doi.org/10.1016/j.chemgeo.2009.07.015, 2009.
Eiler, J. M.: Paleoclimate reconstruction using carbonate clumped isotope thermometry, Quaternary. Sci. Rev., 30, 3575–3588, https://doi.org/10.1016/j.quascirev.2011.09.001, 2011.
Falk, E. S., Guo, W., Paukert, A. N., Matter, J. M., Mervine, E. M., and Kelemen, P. B.: Controls on the stable isotope compositions of travertine from hyperalkaline springs in Oman: Insights from clumped isotope measurements, Geochim. Cosmochim. Ac., 192, 1–28, https://doi.org/10.1016/j.gca.2016.06.026, 2016.
Fernandez, A., Müller, I. A., Rodríguez-Sanz, L., van Dijk, J., Looser, N., and Bernasconi, S. M.: A reassessment of the precision of carbonate clumped isotope measurements: Implications for calibrations and paleoclimate reconstructions, Geochem. Geophy. Geosy., 18, 4375–4386, https://doi.org/10.1002/2017GC007106, 2017.
Fiebig, J., Bajnai, D., Löffler, N., Methner, K., Krsnik, E., Mulch, A., and Hofmann, S.: Combined high-precision Δ48 and Δ47 analysis of carbonates, Chem. Geol., 522, 186–191, https://doi.org/10.1016/j.chemgeo.2019.05.019, 2019.
Ghosh, P., Adkins, J., Affek, H., Balta, B., Guo, W., Schauble, E., Schrag, D., and Eiler, J.: 13C–18O bonds in carbonate minerals: a new kind of paleothermometer, Geochim. Cosmochim. Ac., 70, 1439–1456, https://doi.org/10.1016/j.gca.2005.11.014, 2006.
Green, M. and Taube, H.: Isotopic fractionation in the OH–H2O exchange reaction, J. Phys. Chem., 67, 1565–1566, https://doi.org/10.1021/j100801a507, 1963.
Guo, W.: Carbonate clumped isotope thermometry: application to carbonaceous chondrites and effects of kinetic isotope fractionation, PhD thesis, California Institute of Technology, 261 pp., 2009.
Guo, W., Mosenfelder, J. L., Goddard III, W. A., and Eiler, J. M.: Isotopic fractionations associated with phosphoric acid digestion of carbonate minerals: insights from first principles theoretical modeling and clumped isotope measurements, Geochim. Cosmichim. Ac., 73, 7203–7225, https://doi.org/10.1016/j.gca.2009.05.071, 2009.
Henkes, G. A., Passey, B. H., Grossman, E. L., Shenton, B. J., Yancey, T. E., and Pérez-Huerta, A.: Temperature evolution and the oxygen isotope composition of Phanerozoic oceans from carbonate clumped isotope thermometry, Earth Planet. Sc. Lett., 490, 40–50, https://doi.org/10.1016/j.epsl.2018.02.001, 2018.
Hermoso, M., Lefeuvre, B., Minoletti, F., and de Rafélis, M.: Extreme strontium concentrations reveal specific biomineralization pathways in certain coccolithophores with implications for the Sr/Ca paleoproductivity proxy, PLoS ONE, 12, e0185655, https://doi.org/10.1371/journal.pone.0185655, 2017.
Katz, A., Bonifacie, M., Hermoso, M., Cartigny, P., and Calmels, D.: Laboratory-grown coccoliths exhibit no vital effect in clumped isotope (Δ47) composition on a range of geologically relevant temperatures, Geochim. Cosmochim. Ac., 208, 335–353, https://doi.org/10.1016/j.gca.2017.02.025, 2017.
Kelson, J. R., Huntington, K. W., Schauer, A. J., Saenger, C., and Lechler, A. R.: Toward a universal carbonate clumped isotope calibration: Diverse synthesis and preparatory methods suggest a single temperature relationship, Geochim. Cosmochim. Ac., 197, 104–131, https://doi.org/10.1016/j.gca.2016.10.010, 2017.
Kim, S. T. and O'Neil, J. R.: Equilibrium and nonequilibrium oxygen isotope effects in synthetic carbonates, Geochim. Cosmochim. Ac., 61, 3461–3475, https://doi.org/10.1016/S0016-7037(97)00169-5, 1997.
Kluge, T., Affek, H. P., Dublyansky, Y., and Spötl, C.: Devils Hole paleotemperatures and implications for oxygen isotope equilibrium fractionation, Earth Planet. Sc. Lett., 400, 251–260, https://doi.org/10.1016/j.epsl.2014.05.047, 2014.
Krajewska, B.: Ureases I. Functional, catalytic and kinetic properties: A review, J. Mol. Catal. B-Enzym., 59, 9–21, https://doi.org/10.1016/j.molcatb.2009.01.003, 2009.
Krebs, H. A. and Roughton, F. J. W.: Carbonic anhydrase as a tool in studying the mechanism of reactions involving H2CO3, CO2 or , Biochem. J., 43, 550, https://doi.org/10.1042/bj0430550, 1948.
Létolle, R., Gegout, P., Gaveau, B., and Moranville-Regourd, M.: Isotope fractionation of 18O during precipitation of carbonates at very high pH, CR Acad. Sci. II, 310, 547–552, 1990.
Liu, Y. W., Eagle, R. A., Aciego, S. M., Gilmore, R. E., and Ries, J. B.: A coastal coccolithophore maintains pH homeostasis and switches carbon sources in response to ocean acidification, Nat. Commun., 9, 2857, https://doi.org/10.1038/s41467-018-04463-7, 2018.
Loyd, S. J., Sample, J., Tripati, R. E., Defliese, W. F., Brooks, K., Hovland, T. M., Marlow, J., Hancock, L. G., Martin, R., and Lyons, T.: Methane seep carbonates yield clumped isotope signatures out of equilibrium with formation temperatures, Nat. Commun., 7, 12274, https://doi.org/10.1038/ncomms12274, 2016.
Mangenot, X., Bonifacie, M., Gasparrini, M., Götz, A., Chaduteau, C., Ader, M., and Rouchon, V.: Coupling Δ47 and fluid inclusion thermometry on carbonate cements to precisely reconstruct the temperature, salinity and δ18O of paleo-groundwater in sedimentary basins, Chem. Geol., 472, 44–57, https://doi.org/10.1016/j.chemgeo.2017.10.011, 2017.
Mangenot, X., Gasparrini, M., Rouchon, V., and Bonifacie, M.: Basin-scale thermal and fluid-flow histories revealed by clumped isotope (Δ47) – Middle Jurassic reservoirs of the Paris Basin, Sedimentology, 65, 123–150, https://doi.org/10.1111/sed.12427, 2018a.
Mangenot, X., Gerdes, A., Gasparrini, M., Bonifacie, M., and Rouchon, V.: An emerging thermo-chronometer for carbonate-bearing rocks: Δ47/(U-Pb), Geology, 46, 1067–1070, https://doi.org/10.1130/G45196.1, 2018b.
Matsuzaki, Y., Yamada, H., Chowdhury, F. A., Higashii, T., Kazama, S., and Onoda, M.: Ab initio study of CO2 capture mechanisms in monoethanolamine aqueous solution: reaction pathways from carbamate to bicarbonate, Enrgy. Proced., 37, 400–406, https://doi.org/10.1016/j.egypro.2013.05.124, 2013.
Milliman, J. D.: Production and accumulation of calcium carbonate in the ocean: budget of a nonsteady state, Global Biogeochem. Cy., 7, 927–957, https://doi.org/10.1029/93GB02524, 1993.
Millo, C., Dupraz, S., Ader, M., Guyot, F., Thaler, C., Foy, E., and Ménez, B.: Carbon isotope fractionation during calcium carbonate precipitation induced by ureolytic bacteria, Geochim. Cosmochim. Ac., 98, 107–124, https://doi.org/10.1016/j.gca.2012.08.029, 2012.
Mobley, H. L. and Hausinger, R. P.: Microbial ureases: significance, regulation, and molecular characterization, Microbiol. Mol. Biol. R., 53, 85–108, 1989.
Outka, D. E. and Williams, D. C.: Sequential coccolith morphogenesis in Hymenomonas carterae, J. Protozool., 18, 285–297, https://doi.org/10.1111/j.1550-7408.1971.tb03319.x, 1971.
Rollion-Bard, C., Chaussidon, M., and France-Lanord, C.: pH control on oxygen isotopic composition of symbiotic corals, Earth Planet. Sc. Lett., 215, 275–288, https://doi.org/10.1016/S0012-821X(03)00391-1, 2003.
Saenger, C., Affek, H. P., Felis, T., Thiagarajan, N., Lough, J. M., and Holcomb, M.: Carbonate clumped isotope variability in shallow water corals: Temperature dependence and growth-related vital effects, Geochim. Cosmochim. Ac., 99, 224–242, https://doi.org/10.1016/j.gca.2012.09.035, 2012.
Schauer, A. J., Kelson, J. R., Saenger, C., and Huntington, K. W.: Choice of 17O correction affects clumped isotope (Δ47) values of CO2 measured with mass spectrometry, Rapid Commun. Mass Sp., 30, 2607–2616, https://doi.org/10.1002/rcm.7743, 2016.
Spooner, P. T., Guo, W., Robinson, L. F., Thiagarajan, N., Hendry, K. R., Rosenheim, B. E., and Leng, M. J.: Clumped isotope composition of cold-water corals: A role for vital effects?, Geochim. Cosmochim. Ac., 179, 123–141, https://doi.org/10.1016/j.gca.2016.01.023, 2016.
Staudigel, P. T. and Swart, P. K.: A kinetic difference between 12C- and 13C-bound oxygen exchange rates results in decoupled δ18O and Δ47 values of equilibrating DIC solutions, Geochem. Geophy. Geosy., 19, 2371–2383, https://doi.org/10.1029/2018GC007500, 2018.
Tang, J., Dietzel, M., Fernandez, A., Tripati, A. K., and Rosenheim, B. E.: Evaluation of kinetic effects on clumped isotope fractionation (Δ47) during inorganic calcite precipitation, Geochim. Cosmochim. Ac., 134, 120–136, https://doi.org/10.1016/j.gca.2014.03.005, 2014.
Thaler, C., Millo, C., Ader, M., Chaduteau, C., Guyot, F., and Ménez, B.: Disequilibrium δ18O values in microbial carbonates as a tracer of metabolic production of dissolved inorganic carbon, Geochim. Cosmochim. Ac., 199, 112–129, https://doi.org/10.1016/j.gca.2016.10.051, 2017.
Thiagarajan, N., Adkins, J., and Eiler, J.: Carbonate clumped isotope thermometry of deep-sea corals and implications for vital effects, Geochim. Cosmochim. Ac., 75, 4416–4425, https://doi.org/10.1016/j.gca.2011.05.004, 2011.
Tripati, A. K., Hill, P. S., Eagle, R. A., Mosenfelder, J. L., Tang, J., Schauble, E. A., Eiler, J. M., Zeebee, R. E., Uchikawa, J., Coplen, T. B., Ries, J. B., and Drew, H.: Beyond temperature: Clumped isotope signatures in dissolved inorganic carbon species and the influence of solution chemistry on carbonate mineral composition, Geochim. Cosmochim. Ac., 166, 344–371, https://doi.org/10.1016/j.gca.2015.06.021, 2015.
Urey, H. C., Lowenstam, H. A., Epstein, S., and McKinney, C. R.: Measurement of paleotemperatures and temperatures of the Upper Cretaceous of England, Denmark, and the south-eastern United States, Geol. Soc. Am. Bull., 62, 399–416, https://doi.org/10.1130/0016-7606(1951)62[399:MOPATO]2.0.CO;2, 1951.
Usdowski, E., Michaelis, J., Bottcher, M. E., and Hoefs, J.: Factors for the oxygen isotope equilibrium fractionation between aqueous and gaseous CO2, carbonic-acid, bicarbonate, carbonate, and water (19 ∘C), Z. Phys. Chem., 170, 237–249, 1991.
Watkins, J. M. and Hunt, J. D.: A process-based model for non-equilibrium clumped isotope effects in carbonates, Earth Planet. Sc. Lett., 432, 152–165, https://doi.org/10.1016/j.epsl.2015.09.042, 2015.
Watkins, J. M., Nielsen, L. C., Ryerson, F. J., and DePaolo, D. J.: The influence of kinetics on the oxygen isotope composition of calcium carbonate, Earth Planet. Sc. Lett., 375, 349–360, https://doi.org/10.1016/j.epsl.2013.05.054, 2013.
Zeebe, R. E. and Wolf-Gladrow, D.: CO2 in Seawater: Equilibrium, Kinetics, Isotopes, Elsevier Oceanography Book Series, Vol. 65, 346 pp., Elsevier Science, Amsterdam, 2001.