the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Dec 2020
| 02 Dec 2020
Diversity and distribution of nitrogen fixation genes in the oxygen minimum zones of the world oceans
Bess B. Ward
Diversity and community composition of nitrogen (N) fixing microbes in the three main oxygen minimum zones (OMZs) of the world ocean were investigated using operational taxonomic unit (OTU) analysis of nifH clone libraries. Representatives of three of the four main clusters of nifH genes were detected. Cluster I sequences were most diverse in the surface waters, and the most abundant OTUs were affiliated with Alpha- and Gammaproteobacteria. Cluster II, III, and IV assemblages were most diverse at oxygen-depleted depths, and none of the sequences were closely related to sequences from cultivated organisms. The OTUs were biogeographically distinct for the most part – there was little overlap among regions, between depths, or between cDNA and DNA. In this study of all three OMZ regions, as well as from the few other published reports from individual OMZ sites, the dominance of a few OTUs was commonly observed. This pattern suggests the dynamic response of the components of the overall diverse assemblage to variable environmental conditions. Community composition in most samples was not clearly explained by environmental factors, but the most abundant OTUs were differentially correlated with the obvious variables, temperature, salinity, oxygen, and nitrite concentrations. Only a few cyanobacterial sequences were detected. The prevalence and diversity of microbes that harbor nifH genes in the OMZ regions, where low rates of N fixation are reported, remains an enigma.
- Article
(2979 KB) - Full-text XML
-
Supplement
(67 KB) - BibTeX
- EndNote





Nitrogen fixation is the biological process that introduces new biologically available nitrogen (N) into the ocean and, thus, constrains the overall productivity of large regions of the ocean where N is limiting to primary production. The most abundant and most important diazotrophs in the ocean are cyanobacteria, members of the filamentous genus Trichodesmium and several unicellular genera, including Crocosphaera sp. and the symbiotic genus Candidatus Atelocyanobacterium thalassa (UCYN-A). Although these cyanobacterial species are widespread and have different biogeographical distributions (Moisander et al., 2010), they are restricted to sunlit surface waters, mainly in tropical or subtropical regions.
Because diazotrophs have an ecological advantage in N-depleted waters, and because those conditions occur in the vicinity of oxygen minimum zones, due to the loss of fixed N by denitrification, it has been proposed that N fixation should be favored in regions of the ocean influenced by OMZs (Deutsch et al., 2007). It has also been suggested that the energetic constraints on N fixation might be partially alleviated under reducing, i.e., anoxic, conditions (Großkopf and LaRoche, 2012). In response to these ideas, the search for organisms with the capacity to fix nitrogen has been focused recently in regions of the ocean that contain OMZs. That search usually takes the form of characterizing and quantifying one of the genes involved in the fixation reaction, nifH, which encodes the dinitrogenase reductase enzyme. Diverse nifH assemblages have been reported from the oxygen minimum zone of the eastern tropical South Pacific (Turk-Kubo et al., 2014; Loescher et al., 2016; Fernandez et al., 2011) and the Costa Rica Dome, at the edge of the OMZ in the eastern tropical North Pacific (Cheung et al., 2016). The search for non-cyanobacterial diazotrophs has resulted in the discovery of diverse nifH genes, but they have rarely been associated with significant rates of N fixation (Moisander et al., 2017; Bentzon-Tilia et al., 2015). Thus, the occurrence and diversity of putative diazotrophs in nitrogen-rich aphotic waters remains unexplained.
Here, we report on the distribution and diversity of nifH genes in all three of the world ocean's major OMZs: the two Pacific OMZs, the eastern tropical North (ETNP) and South (ETSP) Pacific, are both highly productive eastern boundary regions. The ETSP is the one of the most productive regions in the world ocean and has an oxygen-depleted layer of about 400 m at its greatest depth. The ETNP is less well ventilated and less productive, with an anoxic layer of more than 700 m. The third major OMZ is the Arabian Sea, which is geographically constrained to the northern Indian Ocean. It experiences an annual monsoon cycle but is permanently and stably stratified with a maximum anoxic layer of about 800 m. Both surface and anoxic depths as well as both DNA and cDNA (i.e., both the presence and expression of the nifH genes) were investigated. The approach used here to investigate diazotroph assemblages is based on clone library analysis of nifH sequences. Next-generation amplicon sequencing would yield greater numbers of sequences, although it might not overcome the primer bias associated with polymerase chain reaction (PCR) and cloning. The strength of the current study is the inclusion of similar data from all three OMZs. By comparing these results to previous studies using the same and other methods, we find robust biogeographical patterns and community structure among the non-cyanobacterial diazotroph assemblages.
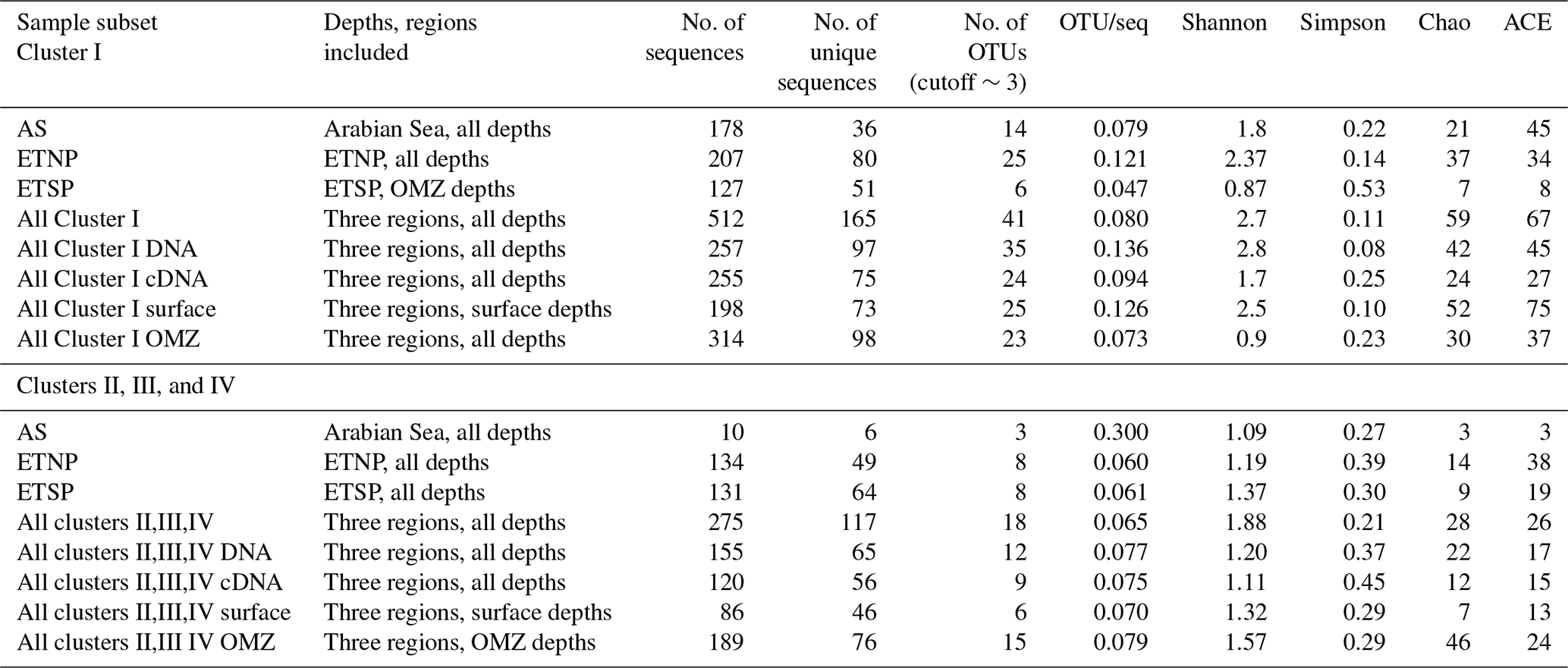
Table 1Sample and clone library descriptions. Sampling regions and depths as well as sequences derived from each depth. S refers to surface (within the euphotic zone); OMZ refers to the oxycline or core of the OMZ, all below the euphotic zone.

Samples analyzed for this study were collected from the three major OMZ regions of the world oceans (16 total samples, Table 1) from the surface and from oxygen minimum zone (OMZ, including oxycline and anoxic) depths. Particulate material from water samples (5–10 L), collected using Niskin samplers mounted on a CTD (Conductivity–Temperature–Depth) rosette system (Sea-Bird Electronics), was filtered onto Sterivex capsules (0.2 µm filter, Millipore, Inc., Bedford, MA) immediately after collection using peristaltic pumps. The filters were flash frozen in liquid nitrogen and stored at −80 ∘C until DNA and RNA could be extracted. For samples from the Arabian Sea, DNA extraction was carried out using the PUREGENE™ Genomic DNA Isolation Kit (Qiagen, Germantown, MD), and the RNA was extracted using the ALLPrep DNA/RNA Mini Kit (Qiagen, Germantown, MD). For samples collected from ETNP and ETSP, DNA and RNA were simultaneously extracted using the ALLPrep DNA/RNA Mini Kit (Qiagen, Germantown, MD). A SuperScript III First Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) was used to synthesize cDNA immediately after extraction following purification of RNA using the procedure described by the manufacturer, including no-RT controls. The extracted DNA was treated with DNase before transcription, and no-RT controls verified the absence of nifH DNA in the RNA preps. DNA was quantified using PicoGreen fluorescence (Molecular Probes, Eugene, OR) calibrated with several dilutions of phage lambda standards.
PCR amplification of nifH genes from environmental sample DNA and cDNA was done on an MJ100 Thermal Cycler (MJ Research) using a Promega PCR kit following the nested reaction (Zehr et al., 1998), with slight modification as in Jayakumar et al. (2017). Briefly, 25 µL PCR reactions containing 50 pmol each of outer primer and 20–25 ng of template DNA were amplified for 30 cycles (1 min at 98 ∘C, 1 min at 57 ∘C, and 1 min at 72 ∘C), followed by amplification with 50 pmol each of inner PCR primers (Zehr and McReynolds, 1989). Water for negative controls and PCR was freshly autoclaved and UV irradiated every day. Negative controls were run with every PCR experiment in order to minimize the possibility of amplifying contaminants (Zehr et al., 2003). The PCR preparation station was also UV irradiated for 1 h before use each day, and the number of amplification cycles was limited to 30 for each reaction. Each reagent was tested separately for amplification in negative controls. nifH bands were excised from PCR products after electrophoresis on 1.2 % agarose gel, and they were cleaned using a QIAquick Nucleotide Removal Kit (Qiagen). Clean nifH products were inserted into a pCR® 2.1-TOPO® vector using One Shot® TOP10 Chemically Competent E. coli and a TOPO TA Cloning® Kit (Invitrogen), according to manufacturer's specifications. This process resulted in 30 clone libraries, 16 of DNA and 14 of RNA, from the 16 samples (Table 1).
Inserted fragments were amplified with M13 Forward (−20) and M13 Reverse primers from randomly picked clones. PCR products were sequenced at Macrogen DNA Analysis Facility using Big Dye™ terminator chemistry (Applied Biosystems, Carlsbad, CA, USA). Sequences were edited using FinchTV version 1.4.0 (Geospiza Inc.), and they were checked for identity using BLAST. Consensus nifH sequences (359 bp) were translated to amino acid (aa) sequences (108 aa after trimming the primer region) and aligned using ClustalW in MEGA X (Kumar et al., 2018; Stecher et al., 2020) along with published nifH sequences from the NCBI database. The alignment was used to construct a maximum likelihood (ML) phylogenetic tree in MEGA X, based on the Poisson model, and the phylogenetic tree was edited using iTOL (Letunic and Bork, 2016). Bootstrap analysis was used to estimate the reliability of phylogenetic reconstruction (1000 iterations). The nifH sequence from Methanosarcina lacustris (AAL02156) was used as an outgroup. The accession numbers from GenBank for the nifH sequences in this study are Arabian Sea DNA sequences JF429940–JF429973 and cDNA sequences accession numbers JQ358610–JQ358707, ETNP DNA sequences KY967751–KY967929 and cDNA sequence KY967930–KY968089, and ETSP DNA sequences MK408165–MK408307 and cDNA sequences MK408308–MK408422.
The nifH nucleotide alignment (of 787 sequences) was used to define operational taxonomic units (OTUs) on the basis of DNA sequence identity. Distance matrices based on this nucleotide alignment were generated in mothur (Schloss and Handlesman, 2009). The relative nifH richness within each clone library was evaluated using rarefaction analysis. OTUs were defined as sequences that differed by ≤ 3 % using the furthest-neighbor method in the mothur program (Schloss and Handlesman, 2009). The 3 % OTU definition is similar to the level at which species are conventionally defined using 16S rDNA sequences, so it may overestimate the meaningful diversity of the functional gene. Redundancy analysis was performed in R using the vegan package. Environmental variables were transformed using decostand.
DNA and cDNA sequences (787 in total) derived from the OMZ regions of the Arabian Sea (AS), the eastern tropical North Pacific (ETNP), and the eastern tropical South Pacific (ETSP) were subjected to OTU and phylogenetic analyses to compare the diversity and community composition, biogeography, and gene expression of nifH-possessing microbes among the three OMZ regions. Phylogenetic analysis of the sequences from the AS, ETNP, and ETSP have been reported separately in previous publications (Jayakumar et al., 2012; Jayakumar et al., 2017; Chang et al., 2019), but the sequences have been combined for additional global analyses here. We compared the threshold OTU definitions at 3 % and 10 % and found that the number of OTUs decreased, as expected, as the resolution decreased. Even at the 3 % threshold, however, OTUs tended to separate by depth and location, indicating a functionally useful distinction at this level. Thresholds of 3 %–5 % as the OTU definition correspond to within and between species-level distinctions for nifH (Gaby et al., 2018). The sequences from the OMZ regions represented three of the four sequence clusters (I, II, III, and IV) described by Zehr et al. (1998).
3.1 Cluster I nifH OTU distributions
Diversity analysis of the nifH Cluster I sequences for the three OMZs based on OTUs using mothur-identified 41 OTUs at a distance threshold of 3 % (Table 2). The number of sequences and the number of OTUs varied widely among depths and stations, so the results are grouped by region (AS, ETNP, and ETSP) or depth horizon (surface or OMZ, including upper oxycline depths), or by cDNA vs. DNA (Table 2). Grouping the sequences by depth horizon (surface or OMZ), region (AS, ETSP, and ETNP), or DNA or RNA, allows for the detection of patterns that are not driven by the relatively low number of sequences obtained from some of the individual clone libraries. The OTUs are numbered in order of decreasing abundance in the clone library, i.e., OTU-1 was the most common OTU; OTU designations for Cluster I are listed in Table 1 in the Supplement.
Table 2OTU Summary. OTU summary for both clusters. Richness and diversity statistics for nifH clone libraries from three OMZ regions. ACE and Chao are nonparametric estimators that predict the total number of OTUs in the original sample.
For all regions and depths combined, the number of OTUs detected (41) was less than the sum of OTUs detected when each region was analyzed separately (45), indicating that there was some overlap of OTUs among regions. However, the overlap was not large. Only 3 of the 12 most abundant OTUs contained sequences from more than one region, and none contained sequences from all three regions (Fig. 1a). When sequences for all three regions were combined, only 4 of the 12 most abundant OTUs contained sequences from both depth horizons (Fig. 1b). Most OTUs represented a single depth, and many represented a single sample. This suggests a pattern of dominance, rather than evenness, in the nifH assemblage. Therefore, deeper sequencing is expected to discover a larger number of rare OTUs, but it might not change the picture that emerges here of a small number of abundant clades. Interestingly, Cheung et al. (2016) reported a similar pattern of dominance based on a larger DNA sequence dataset from only one location. Using 454-pyrosequencing to obtain a similar number of OTUs (37 total) from the Costa Rica Dome, all of the 15 samples investigated by Cheung et al. (2016) were dominated (> 50 %) by one of five major OTUs.
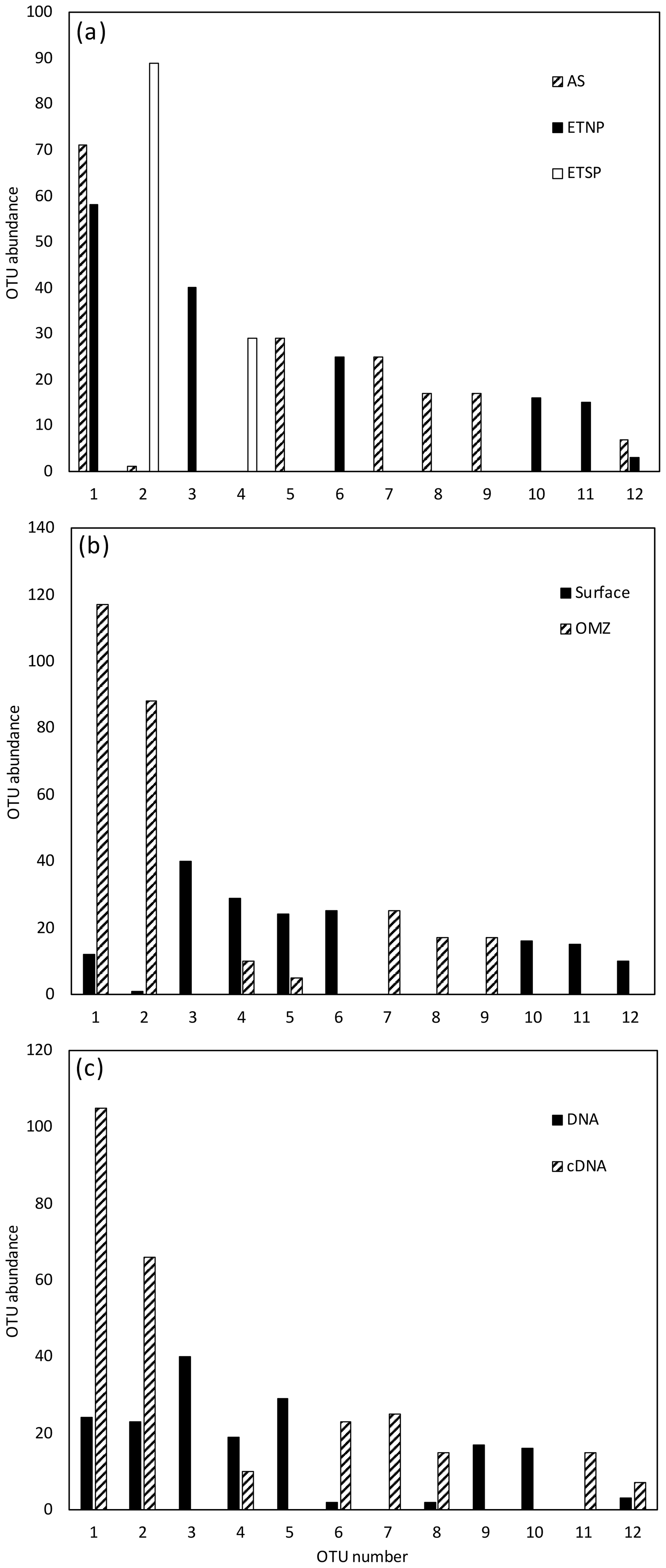
Figure 1Histogram of the 12 most common OTUs from the Cluster I nifH clone libraries from the three OMZ regions. OTUs were considered common if the total number of sequences in an OTU was ≥ 2 % of the total number of nifH clones analyzed (the common OTUs contained 441 of the 512 Cluster I sequences). OTUs were defined according to 3 % nucleotide sequence difference using the furthest-neighbor method. OTU designation is from most common (OTU-1) to least common. (a) OTU distribution among regions. (b) OTU distribution between OMZ (including the core of the ODZ and the upper oxycline depths) and surface depths (oxygenated water). (c) OTU distribution of cDNA vs. DNA clones.
The Arabian Sea was strikingly less diverse than other regions and sample subsets (Fig. 2). For example, when all DNA and cDNA sequences for all depths are grouped together, the Arabian Sea (OTUs = 14, Chao = 21) contains less species richness than the combined surface samples from all three regions (OTUs = 25, Chao = 52), despite having a similar number of total sequences (178 for the Arabian Sea and 198 for all surface samples combined). This lack of diversity in the AS data may be partly due to the preponderance of cDNA sequences, which generally contained less diversity than a similar number of DNA sequences (see below).
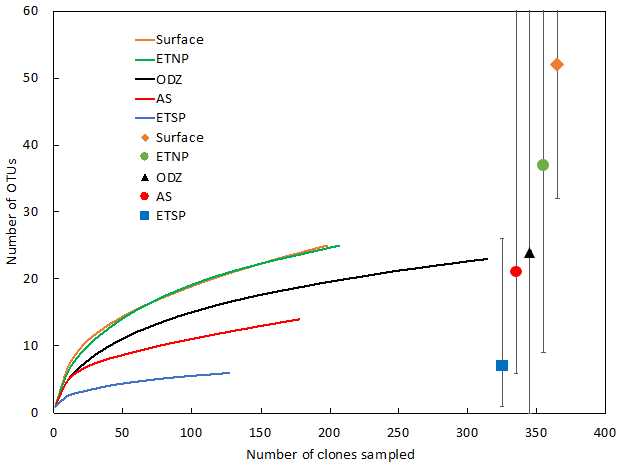
Figure 2Rarefaction curve displaying observed OTU richness vs. the number of clones sequenced for Cluster I nifH sequences (cDNA and DNA). OTUs were defined and designated as in Fig. 1. Chao estimators (individual symbols) are shown for each of the same subsets represented in the rarefaction curves.
Although similar numbers of sequences were obtained for cDNA (255) vs. DNA (257), the OTU “density”, i.e., number of OTUs per number of sequences analyzed, was higher for DNA (0.136 for DNA, 0.094 for cDNA). The Chao statistic verified this observation for the combined data from each region in predicting higher total numbers of OTUs for DNA (Chao = 42) than for cDNA (Chao = 24). This difference could indicate that some of the nifH genes present were not expressed at the time of sampling, but the cDNA sequences were not simply a subset of the DNA community. Half of the 12 most abundant OTUs contained either cDNA or DNA (Fig. 1c), meaning that some genes were never expressed and some expressed genes could not be detected in the DNA. Based on a similar number of sequences from each sample (1–52 per sample) from the ETSP, Turk-Kubo et al. (2014) also found that DNA and cDNA clones were differently distributed among stations; one phylotype was recovered exclusively from cDNA and only one phylotype occurred in both DNA and cDNA. The relatively low sequencing depth associated with clone library studies limits the sensitivity of this comparison, but it clearly shows that dominant components of the DNA and cDNA libraries frequently represent different subsets of the total assemblage.
For all regions combined, similar numbers of OTUs were detected in surface waters (OTUs = 25) and in OMZ samples (OTUs = 23), although a larger number of sequences was analyzed for the OMZ environment (198 vs. 314 sequences for surface and OMZ depths, respectively). It might be expected that the presence of phototrophic diazotrophs in the surface water would lead to greater diversity there, but only one OTU representing a known cyanobacterial phototroph (Katagnymene spiralis or Trichodesmium in OTU-12) was identified, so most of the additional diversity must be present in heterotrophic or unknown sequences.
Rarefaction curves (Fig. 2) indicate that sampling did not approach saturation for region nor depth. The Chao statistic also indicated that much diversity remains to be explored, despite the great uncertainty in these estimates. The total number of OTUs detected, the shape of the rarefaction curve, and the diversity indicators (Fig. 2, Table 2) all indicate that the greatest nifH diversity occurred in surface waters and that much of that diversity was in singletons, i.e., not represented in the 12 most abundant OTUs, which represented 441 (86 %) of the total 512 nifH Cluster I sequences analyzed. Most of that diversity was contained in the ETNP, not solely a function of number of sequences analyzed (Fig. 2).
3.2 Cluster I nifH Phylogeny
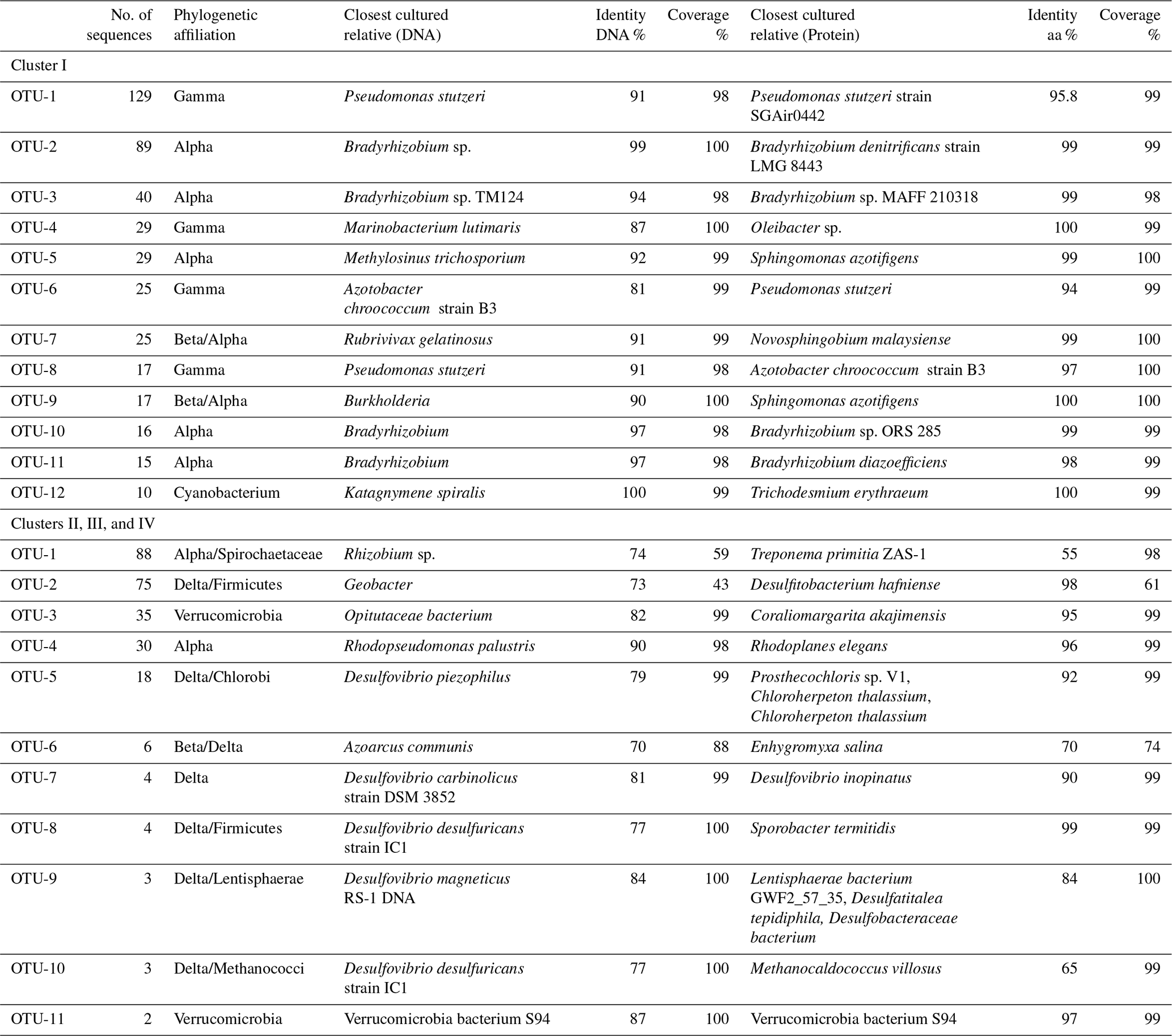
Phylogenetic affiliations at both the DNA and protein level are shown for the 12 most abundant OTUs in Table 3. The most abundant OTU (129 sequences), OTU-1, contained Gammaproteobacterial DNA and cDNA sequences from both the surface and OMZ depths of the ETNP as well as cDNA sequences from oxycline and OMZ depths in the Arabian Sea (Fig. 3). Although very similar to each other, none of these sequences had higher than 91 % identity at the DNA level (96 % at the aa level) with cultivated strains and were most closely related to Pseudomonas stutzeri. P. stutzeri is a commonly isolated marine denitrifier, but it is also known to possess the capacity for N fixation (Krotzky and Werner, 1987). OTU-4, OTU-6, and OTU-8 also contained Gammaproteobacterial sequences. All had high identity with cultivated strains at the protein level, but none were > 91 % identical to cultivated strains at the DNA level.
Table 3OTU identities. OTU identities for both clusters. Cultivated species with closest nucleotide identity to the OTUs identified in the nifH clone libraries from three OMZ regions. Only the 12 most common OTUs (out of 41 total) are listed for Cluster I sequences, and the 11 most common (out of 18 total) are listed for the Cluster II, III, and IV libraries.
Gammaproteobacterial sequences with very close identities to Azotobacter vinelandii have been reported from the Arabian Sea ODZ (oxygen-deficient zone, refers to the depths where oxygen concentrations are low enough to induce anaerobic metabolism, and OMZ denotes the oceanographic region where low-oxygen waters are found) and also from the ETSP (Turk-Kubo et al., 2014). This group of nifH sequences with close identities to A. vinelandii was also retrieved from the English Channel, Himalayan soil, the South Pacific gyre, the Gulf of Mexico, mangrove soil, and many other environments (Fig. 3). Azotobacter-like sequences were included in OTU-6 but were not the closest identity at the DNA level. Although a large number of clones were analyzed here, no sequence that was closely associated with A. vinelandii was retrieved from the three regions. None of the g-244774A11 sequences, Gammaproteobacterial relatives that were abundant in the South Pacific (Moisander et al., 2014), were detected in this study.

Figure 3Maximum likelihood (ML) phylogenetic tree, based on the Poisson model, of Cluster I partial nifH-translated amino acid sequences from DNA and cDNA. Bootstrap values > 50 % of 1000 replications are labeled with black circles on the branches. Accession number of reference sequences from NCBI are provided at the end of each reference name. Positions of the OTUs are shown relative to their nearest neighbors from the database. Individual sequence identities comprising each OTU are listed in Table 3.
OTUs-2, 3, 5, 10, and 11 all represented Alphaproteobacterial sequences, with closest identities to various Bradyrhizobium, Sphingomonas and Methylosinus species. Thus, Alphaproteobacterial sequences (206 sequences) were the most abundant in the clone library. OTU-2 almost exclusively contained ETSP ODZ DNA and cDNA sequences (as well as one AS ODZ DNA sequence). OTU-3 contained DNA sequences from ETNP surface waters. OTU-5 exclusively contained Arabian Sea DNA sequences from Station 3, whereas OTU-10 contained only surface samples from the ETNP. An OTU threshold of 11 % grouped all (179 sequences in five OTUs) of these Alphaproteobacterial sequences together, but the 3 % threshold is consistent with the phylogenetic tree, which shows small-scale biogeographical separation of sequence groups.
OTUs-7 and -9 were identified as Betaproteobacteria with closest identities to Rubrivivax gelatinosus and Burkholderia, 91 % and 90 %, respectively, at the DNA level. However, at the aa level, these sequences were 99 % and 100 % identical to Novosphingobium malaysiense and S. azotifigens, respectively, both Alphaproteobacteria, and they were again biogeographically distinct. OTU-7 contained 25 DNA sequences from the ODZ depths in the Arabian Sea, and OTU-9 contained 17 Burkholderia-like sequences from the oxycline at Station 1 in the Arabian Sea. No Betaproteobacterial nifH sequences were detected in the ETNP or ETSP, but sequences similar to Burkholderia phymatum, Cupriavidus sp., and Sinorhizobium meliloti have previously been reported from the ETSP (Fernandez et al., 2015). Consistent with our previous report, however, there is no clear separation between the alpha and the beta groups in nifH phylogeny (Jayakumar et al., 2017).
Most of the Cluster I ETSP sequences from this study were contained in two OTUs (2 and 4). OTU-2 contained 89 Alphaproteobacterial sequences with > 98 % identity to nifH sequences from Bradyrhizobium sp. Uncultured bacterial sequences retrieved from the South China Sea, the English Channel, mangrove sediment, wastewater treatment, and grassland soil were related to these ETSP sequences. OTU-4 contained 29 Gammaproteobacterial sequences retrieved from both the surface and ODZ depths. Four of the remaining ETSP Cluster I sequences were grouped together as OTU-17 (Alphaproteobacteria, 89 % and 96 % identities with Methyloceanibacter sp. and Bradyrhizobium sp. at the DNA and aa level, respectively), three were in OTU-23 (Bradyrhizobium 100 % identity), and two were singletons. One of the singletons was most closely related to uncultured soil and sediment sequences and to Azorhizobium sp. (86 %) and one had 97 % identity with Bradyrhizobium denitrificans and many sequences from marine sediments.
OTU-22 represents the Deltaproteobacterial group. This novel group has been previously reported from the ETNP (Jayakumar et al., 2017) and has three sequences from the Arabian Sea (OTU-22) and two singletons from ETNP surface waters. nifH-possessing Deltaproteobacteria have been reported not only from all the three ODZs but also in several other marine environments including the Chesapeake Bay water column, microbial mats from an intertidal sandy beach on a Dutch barrier island, Jiaozhou Bay sediment, Rongcheng Bay sediment, the Bohai Sea, the Mediterranean Sea, Narragansett Bay, and the South Pacific gyre.
Proteobacteria-like sequences, especially Alpha- and Gammaproteobacteria, are the most frequently reported nifH sequences from the OMZs studied here and similar environments. A total of 31 of 37 OTUs detected by Cheung et al. (2016) in the Costa Rica Dome OMZ were Proteobacteria, with the two most common OTUs being closely related to Alphaproteobacterium Methylocella palustris and the Gammaproteobacterium Vibrio diazotrophicus. Loescher et al. (2014, 2016) also found V. diazotrophicus-like sequences as well as several other Gammaproteobacteria in the ETSP. V. diazotrophicus has been previously reported in the Arabian Sea (Jayakumar et al., 2012) but was not prominent in the present study. Sequences most similar to various V. diazotrophicus, other Vibrio species, and other Gammaproteobacteria, including P. stutzeri, were the most common non-cyanobacterial Cluster I sequences reported for the low-oxygen waters of the Southern California Bight (Hamersley et al., 2011). Bradyrhizobium spp., one of the most common genera reported here and in surface waters of the Arabian Sea (Bird and Wyman, 2013) as well as by Fernandez et al. (2011) in the ETSP were also detected in the Costa Rica Dome OMZ and were the dominant OTU at 1000 m at one station (Cheung et al., 2016). Bradyrhizobium-like sequences were the most abundant among those amplified from ODZ incubations in which the N2 fixation rate was enhanced by the addition of glucose (Bonnet et al., 2013). In addition to Bradyrhizobium-like and Teredinibacter-like nifH sequences, Turk-Kubo et al. (2014) found four other abundant Gammaproteobacteria-like nifH sequences, which were entirely novel. The “Gamma A”, which are commonly reported non-cyanobacteria diazotroph nifH sequences from non-OMZ environments (Langlois et al., 2015; Moisander et al., 2017), were represented by a singleton from the ETNP in the present study.
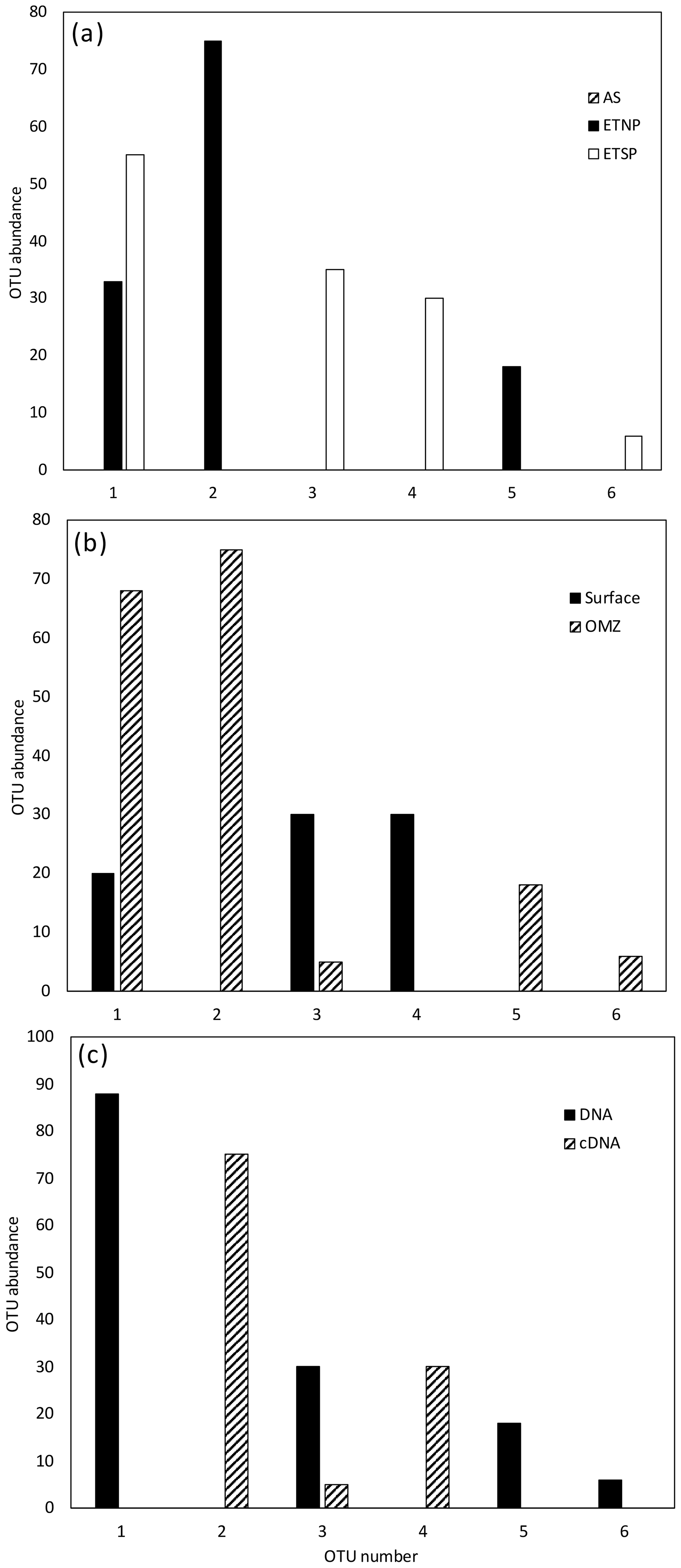
Figure 4Histogram of the six most common OTUs from the Cluster II, III, and IV nifH clone libraries from the three OMZ regions. OTUs were considered common if the total number of sequences in an OTU was ≥ 2 % of the total number of nifH clones analyzed (the common OTUs contained 252 of the 275 Cluster II, III, and IV sequences). OTUs were defined according to 3 % nucleotide sequence difference using the furthest-neighbor method. OTU designation is from most common (OTU-1) to least common. (a) OTU distribution among regions. (b) OTU distribution between OMZ (including core of the ODZ and the upper oxycline depths) and surface depths (oxygenated water). (c) OTU distribution of cDNA vs. DNA clones.

Figure 5Rarefaction curve displaying observed OTU richness vs. the number of clones sequenced for Cluster II, III, and IV nifH sequences (cDNA and DNA). OTUs were defined and designated as in Fig. 4. Chao estimators (individual symbols) are shown for each of the same subsets represented in the rarefaction curves.
nifH sequences related to various Alphaproteobacterial methylotrophs are commonly found in OMZs: Methylosinus trichosporium-like sequences, which are reported here in OTU-5 from the Arabian Sea both at the surface and at ODZ depths, were also reported by Fernandez et al. (2011) in the ETSP. Methylocella palustris-like nifH genes comprised the most common OTU in the ODZ core depths in the Costa Rica Dome (Cheung et al., 2016). M trichosporium and M. palustris represent obligate and facultative methanotrophs, respectively, and are both also obligately aerobic. Detection of nifH genes closely related to those of methanotrophs does not prove that methanotrophy is present or important in the anoxic environment of the ODZ, but the consistency of this finding across sites motivates further investigation of the potential for methane production and consumption in ODZs.
The pattern of the high diversity of nifH-bearing, mostly heterotrophic microbes, in addition to the dominance of one or a small number of nifH OTUs in each sample, suggests a bloom and bust pattern of organic matter-supported growth. That is, we suggest that organic matter, which is supplied episodically in the upwelling regimes, stimulates the growth of copiotrophic microbes that respond rapidly in a bloom-like fashion. This bloom scenario has been described for denitrifying bacteria based on the OTU patterns observed in the nirS and nirK genes as a function of the stage of denitrification in both natural assemblages and incubated samples from OMZs (Jayakumar et al., 2009). Amino acids and glucose both stimulated N2 fixation in OMZ samples from the ETSP, and nifH sequences associated with Alpha- and Gammaproteobacteria, as well as Cluster III phylotypes, were found in a glucose enrichment experiment (Bonnet et al., 2013) The role of nifH in these heterotrophic microbes is unclear, especially because rates of nitrogen fixation in these locations in the absence of cyanobacteria or nutrient enrichment is often very low (Turk-Kubo et al., 2014; Loescher et al., 2016; Chang et al., 2019).
Although Trichodesmium-like clones have been retrieved from the surface waters of the Arabian Sea and the ETNP OMZs, only 10 clones (OTU-12) in the combined clone library analyzed here were related to Trichodesmium (98 % identity), including both cDNA and DNA from the Arabian Sea and cDNA from the ETNP. These sequences were actually 100 % identical to Katagnymene spiralis, a close relative of Trichodesmium isolated from the South Pacific Ocean. Turk-Kubo et al. (2014) also retrieved only a few cyanobacterial sequences from the ETSP. No other cyanobacterial nifH sequences were identified.
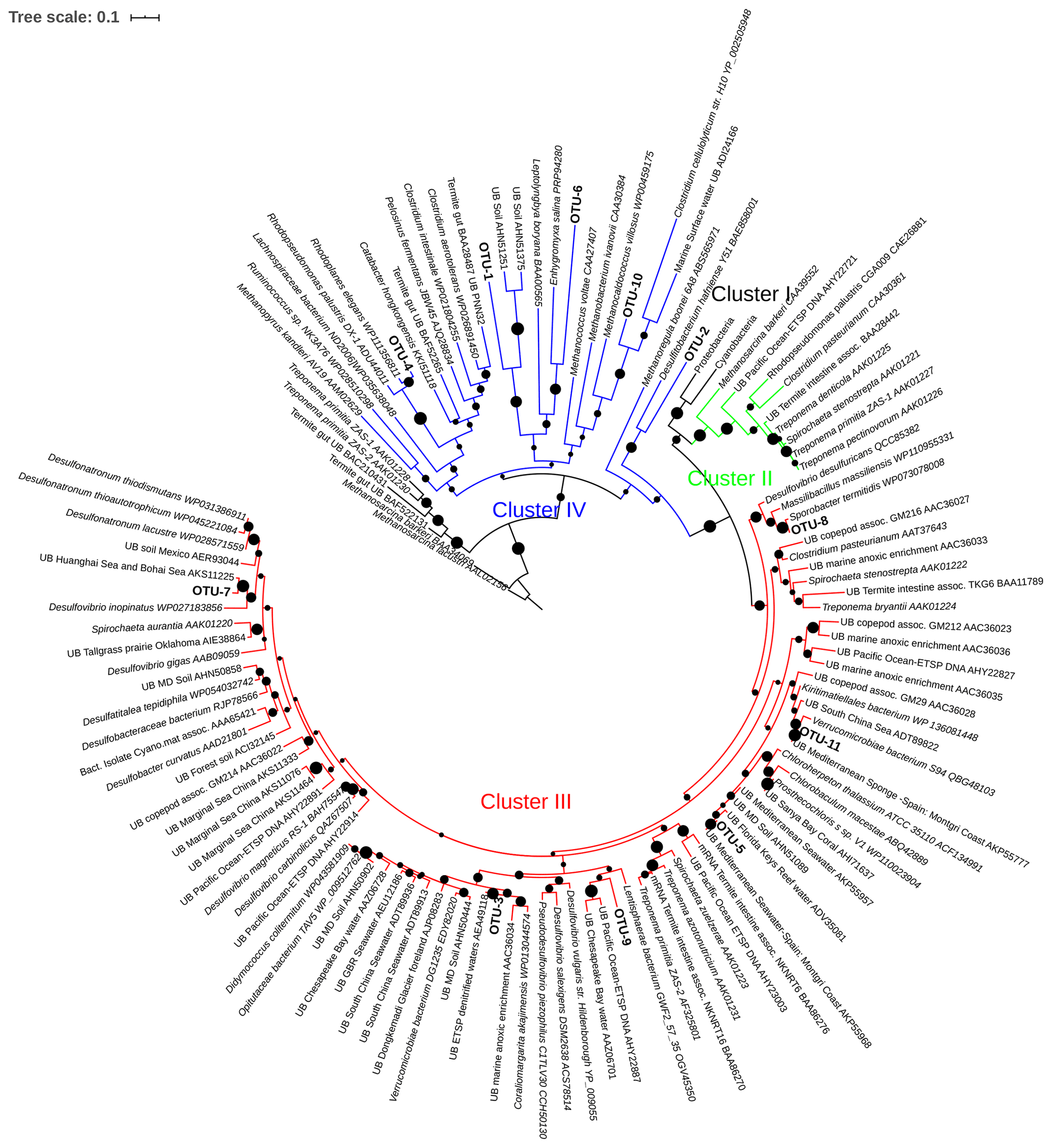
Figure 6Maximum likelihood (ML) phylogenetic tree, based on the Poisson model, of Cluster II, III, and IV partial nifH-translated amino acid sequences from DNA and cDNA. Bootstrap values > 50 % of 1000 replications are labeled with black circles on the branches. Accession number of reference sequences from NCBI are provided at the end of each reference name. Positions of the OTUs are shown relative to their nearest neighbors from the database. Individual sequence identities comprising each OTU are listed in Table 3.
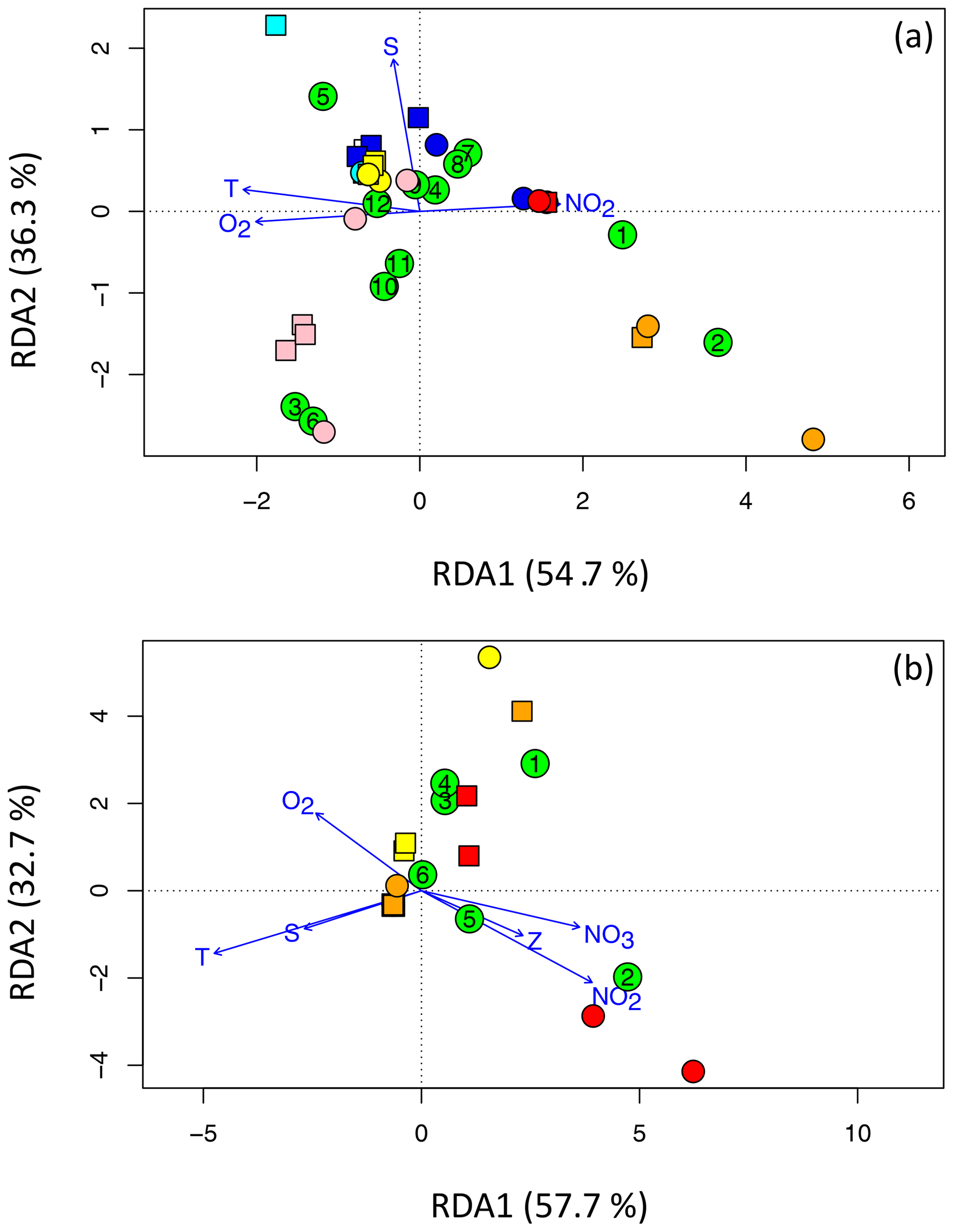
Figure 7RDA plots for (a) Cluster I and (b) clusters II, III, and IV, illustrating the relationships among OTUs (green circles containing the OTU number) and sites. DNA is represented using squares, and cDNA is represented using circles. The Arabian Sea is cyan (surface) and blue (OMZ), the ETNP is pink (surface) and red (deep), and the ETSP is yellow (surface) and orange (deep). Panel (a) shows the 12 most abundant OTUs for Cluster I and the four most independent environmental variables, T denotes temperature, S denotes salinity, NO2 denotes the nitrite concentration, and O2 denotes the oxygen concentration. Panel (b) shows the six most abundant OTUs for clusters II, III, and IV and all six environmental variables, NO3 denotes the nitrate concentration and Z denotes depth.
3.3 Clusters II, III, IV nifH OTU distributions
The other three nifH clusters were combined for OTU analysis due to the limited number of sequences and OTUs obtained. A total of 18 OTUs were identified in the combined set of 275 sequences with a 3 % distance threshold (Table 2); OTU designations for Cluster II, III, and IV are listed in Table 2 in the Supplement. Most of the Cluster II, III, and IV sequences were from the ETNP and ETSP. As with the Cluster I sequences, there was very little geographic and depth overlap among these OTUs (Fig. 4a, b). Only OTU-1 contained sequences from more than one site, the ETNP and the ETSP. OTU-2 contained only cDNA sequences representing ODZ depths at both ETNP stations. OTU-3 exclusively contained ETSP DNA sequences from the surface and cDNA sequences from ODZ depths. Only 10 of the Cluster II, III, and IV sequences were from the Arabian Sea, and they formed three separate OTUs, a greater “OTU density” than was present at either of the Pacific sites. As observed for Cluster I, most of the OTUs that were detected in the DNA were not being expressed, and those that were expressed were not detected in the DNA (Fig. 4c).
Rarefaction curves (Fig. 5) indicate that sampling for Cluster II, III, and IV did not approach saturation. The Chao statistic also indicated that much diversity remains to be explored, despite the great uncertainty in these estimates. Unlike the Cluster I analysis, there were relatively few singletons in the Cluster II, III, and IV data, and the assemblages were dominated by a few types.
3.4 Cluster II, III, and IV nifH phylogeny
Four large OTUs (OTU-1, -2, -4, and -6) in clusters II, III, and IV belonged to nifH Cluster IV, and Alphaproteobacteria/Spirochaeta and Deltaproteobacteria were the dominant phylogenies (Table 3, Fig. 6). The largest OTU, OTU-1, contained 88 DNA sequences from the ETNP ODZ depths from both stations and from both depths in the ETSP. This OTU had no similarity to any cultured microbe. OTU-4 contained 30 sequences from the ETSP, all cDNA from one surface station, in nifH Cluster IV.
OTU-2 (75 sequences) in Cluster IV contained only cDNA sequences, all from ODZ samples in the ETNP (both stations), and had no close relatives among cultivated species. Although Turk-Kubo et al. (2014) retrieved a few clones identified as belonging to Cluster II from the euphotic zone of the ETSP, we did not find any sequence falling into this cluster. OTU-3 contained 35 sequences in Cluster III and was dominated by DNA sequences from surface depths of the ETSP. OTU-5 represented Deltaproteobacteria in nifH Cluster III and contained 18 identical DNA sequences from 90 m at Station BB1 in the ETNP. Thus, of the five most common OTUs (89 % of the total Cluster II, III, and IV sequences analyzed), only one could be identified as a closely related genus (i.e., OTU-4 with 90 % identity with R. palustris) and there was no overlap between DNA and cDNA OTUs from the same depths.
The other 13 OTUs in the Cluster II, III, and IV sequences represented either Cluster III or IV. None of these were very closely related to any cultivated sequences. OTU-6 contained both DNA and cDNA from the OMZ at one ETSP station. OTU-7 contained four sequences from ETNP surface waters with close identities to a sequence retrieved from the Bohai Sea. OTU-11 had one DNA and one cDNA sequences from the ETSP. All of the other sequences were less than 84 % identical to any sequence in the database and could only be loosely identified as Firmicutes or Proteobacteria.
Although there were few high identities with known species, many of the Cluster II, III, and IV sequences (OTUs -2, -5, -7, -9, and -10) were most closely affiliated with sulfate-reducing clades at either the DNA or protein level. Four OTUs with highest identity to known sulfate reducers were reported by Cheung et al. (2016), and one of them comprised nearly 40 % of the sequences in one anoxic sample. nifH sequences that cluster with Desulfovibrio spp. are often reported from ODZ samples (Turk-Kubo et al., 2014; Loescher et al., 2014; Fernandez et al., 2011). Consistent reports of nifH genes associated with obligate anaerobes involved in sulfate reduction suggests a role for this metabolism in the ODZ, again motivating further research on the significance of both sulfate reduction and associated N2 fixation in ODZ waters.
3.5 Biogeography and environmental correlations
The dominant factor determining OTU composition and distribution is clearly biogeography (Fig. 4). That geographical factor is also evident in the redundancy analysis (Fig. 7). (Only sites that contained sequences from one of the top OTUs are represented in the plots, so the number of site symbols is less than 30 for both plots.) For example, Cluster I OTU-5 containing only Arabian Sea surface sequences was positively correlated with both temperature (T) and salinity (S) and all of the Arabian Sea samples clustered in the quadrant associated with high T and S (Fig. 7a). Surface samples from the ETSP were also in that quadrant, but surface ETNP samples were negatively correlated with S. The surface ETNP samples correlated with OTUs-3. -6, -10, and -11, all of which contained exclusively surface samples. The two largest Cluster I OTUs were associated with the deep samples from the ETNP and ETSP and correlated positively with nitrite concentration and negatively with oxygen – a signature of the OMZ. Nitrate concentration and depth did not increase the power of the analysis and were omitted from the Cluster I RDA. Most of the sites and five of the most common Cluster I OTUs were not well differentiated by any of the usual environmental parameters.
The Arabian Sea contained very few sequences in clusters II, III, and IV and none of them were in the top six OTUs, so only ETNP and ETSP samples are represented in the RDA for these clusters (Fig. 7b). The two largest OTUs in clusters II, III, and IV were negatively correlated with T and S but separated along the second RDA axis, demonstrating opposite relationships with oxygen, nitrite, and nitrate concentrations. OTU-1 included ETSP surface sequences, as well as ODZ sequences from both ETNP and ETSP, whereas OTU-2 contained only ODZ sequences but both OTUs were phylogenetically related to anaerobic clades (Table 2). Inclusion of all six environmental variables was necessary to obtain maximum separation of the sites and OTUs for clusters II, III, and IV.
The OMZ regions of the world ocean contain substantial nifH diversity, both in surface waters and at oxygen-depleted intermediate depths. Surface waters contained greater diversity for Cluster I, but the ODZ held the highest diversity for clusters II, III, and IV. Cyanobacterial sequences were rare in the combined dataset and were not detected in the ETSP. The ETSP contained the least diversity of Cluster I sequences, while Cluster II, III, and IV were least abundant and least diverse in clone libraries from the Arabian Sea. Most of the sequences in all three clusters of the conventional nifH phylogeny were not closely related to any sequences from cultivated Bacteria or Archaea. The most abundant OTUs in Cluster I and in clusters II, III, and IV could be assigned to the Alphaproteobacteria, followed by the Gammaproteobacteria for Cluster I and Deltaproteobacteria for Cluster II, III, and IV sequences. Most of the OTUs were not shared among regions, depths, or DNA vs. cDNA and were sometimes restricted to individual samples. Some Cluster I sequences had high identity to known species (e.g., Bradyrhizobium and Trichodesmium), but most of the Cluster II, III, and IV sequences were only distantly related to any cultured species.
The assemblage composition of nifH-bearing microbes is mainly explained by region, but OTU composition was also consistent with the influence of key environmental parameters such as oxygen and temperature, and reflects association with the secondary nitrite maximum for deep samples. There are few studies that report nifH sequences from oceanic OMZs (Jayakumar et al., 2012, from the Arabian Sea; Fernandez et al., 2011; Loescher et al., 2014; Turk-Kubo et al., 2014, all from the ETSP) or similar environments (Cheung et al., 2016, from the Costa Rica Dome and Hamersley et al., 2011, from hypoxic basins in the Southern California Bight). Combining those reports from individual regions, as well as the new sequences from the ETNP reported here, shows that most of the sites and depths, both in this study and in others from OMZ regions, are dominated by one or a few OTUs, which suggests bloom-type dynamics within a diverse background assemblage. Microbes occupying very similar niches and present at low population levels might respond differentially to episodic inputs of organic matter, resulting in spatially and temporally varying dominance by a few clades. Thus, we find nifH sequences associated with similar metabolic types represented across all the OMZs, although the specific species- and genus-level affiliations differ. The consistent detection of nifH sequences related to those found in known sulfate reducers and methanotrophs suggests the need for further investigation of these pathways in ODZs.
While measurements of N2 fixation rates are not reported here, the abundance of cDNA sequences suggests that the cells harboring these genes are active. Low but analytically significant rates have been detected in ODZ depths in the ETNP (Jayakumar et al., 2017) and ETSP (Chang et al., 2019), which suggests that non-cyanobacterial N2 fixation could make a minor contribution to the nitrogen budget of the ocean. Therefore, it is important in future work to determine how the diversity described here actually contributes to biogeochemically significant reactions and what environmental and biotic factors might influence or control the activity of diazotrophs in the OMZ.
The primary sequence data have been deposited at GenBank using the accession numbers listed in Sect. 2.
Supplement Table S1: List of sequences included in each OTU for Cluster I nifH sequences. Supplement Table S2: List of sequences included in each OTU for Cluster II, III, IV nifH sequences The supplement related to this article is available online at: https://doi.org/10.5194/bg-17-5953-2020-supplement.
AJ performed the experiments and phylogenetic analysis, BBW performed the statistical analysis, and AJ and BBW wrote the paper.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Ocean deoxygenation: drivers and consequences – past, present and future (BG/CP/OS inter-journal SI)”. It is a result of the International Conference on Ocean Deoxygenation, Kiel, Germany, 3–7 September 2018.
The authors are grateful for the help provided by the captain and crew of R/V Roger Revelle, Thomas G. Thompson and Nathanial B. Palmer. We thank all the anonymous reviewers for their constructive comments that improved this paper immensely.
This research has been supported by the NSF (grant no. OCE-1356043 and OCE-1029951).
This paper was edited by Anja Engel and reviewed by three anonymous referees.
Bentzon-Tilia, M., Traving, S. J., Mantikci, M., Knudsen-Leerbeck, H., Hansen, J. L. S., Markager, S., and Riemann, L.: Significant N-2 fixation by heterotrophs, photoheterotrophs and heterocystous cyanobacteria in two temperate estuaries, Isme J., 9, 273–285, https://doi.org/10.1038/ismej.2014.119, 2015.
Bird, C. and Wyman, M.: Transcriptionally active heterotrophic diazotrophs are widespread in the upper water column of the Arabian Sea, Fems Microbiol. Ecol., 84, 189–200, https://doi.org/10.1111/1574-6941.12049, 2013.
Bonnet, S., Dekaezemacker, J., Turk-Kubo, K. A., Moutin, T., Hamersley, R. M., Grosso, O., Zehr, J. P., and Capone, D. G.: Aphotic N-2 Fixation in the Eastern Tropical South Pacific Ocean, Plos One, 8, e81265, https://doi.org/10.1371/journal.pone.0081265, 2013.
Chang, B. X., Jayakumar, A., Widner, B., Bernhardt, P., Mordy, C. M., Mulholland, M. R., and Ward, B. B.: Low rates of dinitrogen fixation in the eastern tropical South Pacific, Limnol. Oceanogr., 64, 1913–1923, https://doi.org/10.1002/lno.11159, 2019.
Cheung, S. Y., Xia, X. M., Guoand, C., and Liu, H. B.: Diazotroph community structure in the deep oxygen minimum zone of the Costa Rica Dome, J. Plankton Res., 38, 380–391, https://doi.org/10.1093/plankt/fbw003, 2016.
Deutsch, C., Sarmiento, J. L., Sigman, D. M., Gruber, N., and Dunne, J. P.: Spatial coupling of nitrogen inputs and losses in the ocean, Nature, 445, 163–167, 2007.
Fernandez, C., Farias, L., and Ulloa, O.: Nitrogen Fixation in Denitrified Marine Waters, Plos One, 6, e20539, https://doi.org/10.1371/journal.pone.0020539, 2011.
Fernandez, C., Lorena Gonzalez, M., Munoz, C., Molina, V., and Farias, L.: Temporal and spatial variability of biological nitrogen fixation off the upwelling system of central Chile (35–38.5∘ S), J. Geophys. Res.-Ocean., 120, 3330–3349, https://doi.org/10.1002/2014jc010410, 2015.
Gaby, J. C., Rishishwar, L., Valderrama-Aguirre, L. C., Green, S. J., Valderrama-Aguirre, A., Jordan, I. L., and Kostka, J. E.: Diazotroph community characterization via a high-throughput nifH amplicon sequencing and analysis pipeline, Appl. Environ. Microbiol., 84, 1512–01517, https://doi.org/10.1128/AEM.01512-17, 2018.
Großkopf, T. and LaRoche, J.: Direct and indirect costs of dinitrogen fixation in Crocosphaera watsonii WH8501 and possible implications for the nitrogen cycle, Front. Microbiol., 3, 236, https://doi.org/10.3389/fmicb.2012.00236, 2012.
Hamersley, M. R., Turk, K. A., Leinweber, A., Gruber, N., Zehr, J. P., Gunderson, T., and Capone, D. G.: Nitrogen fixation within the water column associated with two hypoxic basins in the Southern California Bight, Aquat. Microb. Ecol., 63, 193–205, 2011.
Jayakumar, A., O'Mullan, G. D., Naqvi, S. W. A., and Ward, B. B.: Denitrifying bacterial community composition changes associated with stages of denitrification in oxygen minimum zones, Microbiol. Ecol., 58, 350–362, 2009.
Jayakumar, A., Al-Rshaidat, M. M. D., Ward, B. B., and Mulholland, M. R.: Diversity, distribution, and expression of diazotroph nifH genes in oxygen-deficient waters of the Arabian Sea, Fems Microbiol. Ecol., 82, 597–606, 2012.
Jayakumar, A., Chang, B. N. X., Widner, B., Bernhardt, P., Mulholland, M. R., and Ward, B. B.: Biological nitrogen fixation in the oxygen-minimum region of the eastern tropical North Pacific ocean, Isme J., 11, 2356–2367, https://doi.org/10.1038/ismej.2017.97, 2017.
Krotzky, A. and Werner, D.: Nitrogen fixation in Pseudomonas stutzeri, Arch. Microbiol., 147, 48–57, https://doi.org/10.1007/bf00492904, 1987.
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K.: MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms, Mol. Biol. Evol., 35, 1547–1549, https://doi.org/10.1093/molbev/msy096, 2018.
Langlois, R., Grokopf, T., Mills, M., Takeda, S., and LaRoche, J.: Widespread Distribution and Expression of Gamma A (UMB), an Uncultured, Diazotrophic, gamma-Proteobacterial nifH Phylotype, Plos One, 10, e0128912, https://doi.org/10.1371/journal.pone.0128912, 2015.
Letunic, I. and Bork, P.: Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees, Nucl. Acids Res., 44, W242–W245, https://doi.org/10.1093/nar/gkw290, 2016.
Loescher, C. R., Grosskopf, T., Desai, F. D., Gill, D., Schunck, H., Croot, P. L., Schlosser, C., Neulinger, S. C., Pinnow, N., Lavik, G., Kuypers, M. M. M., LaRoche, J., and Schmitz, R. A.: Facets of diazotrophy in the oxygen minimum zone waters off Peru, Isme J., 8, 2180–2192, https://doi.org/10.1038/ismej.2014.71, 2014.
Loescher, C. R., Bange, H. W., Schmitz, R. A., Callbeck, C. M., Engel, A., Hauss, H., Kanzow, T., Kiko, R., Lavik, G., Loginova, A., Melzner, F., Meyer, J., Neulinger, S. C., Pahlow, M., Riebesell, U., Schunck, H., Thomsen, S., and Wagner, H.: Water column biogeochemistry of oxygen minimum zones in the eastern tropical North Atlantic and eastern tropical South Pacific oceans, Biogeosciences, 13, 3585–3606, https://doi.org/10.5194/bg-13-3585-2016, 2016.
Moisander, P. H., Beinart, R. A., Hewson, I., White, A. E., Johnson, K. S., Carlson, C. A., Montoya, J. P., and Zehr, J. P.: Unicellular Cyanobacterial Distributions Broaden the Oceanic N-2 Fixation Domain, Science, 327, 1512–1514, https://doi.org/10.1126/science.1185468, 2010.
Moisander, P. H., Serros, T., Paerl, R. W., Beinart, R. A., and Zehr, J. P.: Gammaproteobacterial diazotrophs and nifH gene expression in surface waters of the South Pacific Ocean, ISME J., 8, 1962–1973, 2014.
Moisander, P. H., Benavides, M., Bonnet, S., Berman-Frank, I., White, A. E., and Riemann, L.: Chasing after Non-cyanobacterial Nitrogen Fixation in Marine Pelagic Environments, Front. Microbiol., 8, 1736, https://doi.org/10.3389/fmicb.2017.01736, 2017.
Schloss, P. D. and Handlesman, J.: Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness, Appl. Environ. Microbiol., 71, 1501–1506, 2009.
Stecher, G., Tamura, K., and Kumar, S.: Molecular Evolutionary Genetics Analysis (MEGA) for macOS, Mol. Biol. Evol., 37, 1237–1239, https://doi.org/10.1093/molbev/msz312, 2020.
Turk-Kubo, K. A., Karamchandani, M., Capone, D. G., and Zehr, J. P.: The paradox of marine heterotrophic nitrogen fixation: abundances of heterotrophic diazotrophs do not account for nitrogen fixation rates in the Eastern Tropical South Pacific, Environ. Microbiol., 16, 3095–3114, https://doi.org/10.1111/1462-2920.12346, 2014.
Zehr, J. P. and McReynolds, L. A.: Use of degenerate oligonucleotides for amplification of the nifH gene from the marine cyanobacterium Trichodesmium theiebautii, Appl. Environ. Microbiol., 55, 2522–2526, 1989.
Zehr, J. P., Mellon, M. T., and Zani, S.: New nitrogen-fixing microorganisms detected in oligotrophic oceans by amplification of nitrogenase (nifH) genes, Appl. Environ. Microbiol., 6, 3444–3450, 1998.
Zehr, J. P., Crumbliss, L. L., Church, M. J., Omoregie, E. O., and Jenkins, B. D.: Nitrogenase genes in PCR and RT-PCR reagents: implications for studies of diversity of functional genes, Biotechniques, 35, 996–1005, 2003.