the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Feb 2021
| 16 Feb 2021
Age distribution, extractability, and stability of mineral-bound organic carbon in central European soils
Marion Schrumpf
Klaus Kaiser
Allegra Mayer
Günter Hempel
Susan Trumbore
The largest share of total soil organic carbon (OC) is associated with minerals. However, the factors that determine the amount and turnover of slower- versus faster-cycling components of mineral-associated carbon (MOC) are still poorly understood. Bioavailability of MOC is thought to be regulated by desorption, which can be facilitated by displacement and mobilization by competing ions. However, MOC stability is usually determined by exposure to chemical oxidation, which addresses the chemical stability of the organic compounds rather than the bonding strength of the OC–mineral bond. We used a solution of NaOH, a strong agent for desorption due to high pH, and NaF, adding F−, a strongly sorbing anion that can replace anionic organic molecules on mineral surfaces, to measure the maximum potentially desorbable MOC. For comparison, we measured maximal potential oxidation of MOC using heated H2O2. We selected MOC samples (> 1.6 g cm3) obtained from density fractionation of samples from three soil depth increments (0–5, 10–20, and 30–40 cm) of five typical soils of central Europe, with a range of clay and pedogenic oxide contents, and under different ecosystem types (one coniferous forest, two deciduous forests, one grassland, and one cropland). Extracts and residues were analysed for OC and 14C contents, and further chemically characterized by cross-polarization magic angle spinning 13C-nuclear magnetic resonance (CPMAS-13C-NMR). We expected that NaF–NaOH extraction would remove less and younger MOC than H2O2 oxidation and that the NaF–NaOH extractability of MOC is reduced in subsoils and soils with high pedogenic oxide contents.
The results showed that a surprisingly consistent proportion of 58 ± 11 % (standard deviation) of MOC was extracted with NaF–NaOH across soils, independent of depth, mineral assemblage, or land use conditions. NMR spectra revealed strong similarities in the extracted organic matter, with more than 80 % of OC in the O/N (oxygen and/or nitrogen) alkyl and alkyl C region. Total MOC amounts were correlated with the content of pedogenic oxides across sites, independent of variations in total clay, and the same was true for OC in extraction residues. Thus, the uniform extractability of MOC may be explained by dominant interactions between OC and pedogenic oxides across all study sites. While Δ14C values of bulk MOC suggested differences in OC turnover between sites, these were not linked to differences in MOC extractability. As expected, OC contents of residues had more negative Δ14C values than extracts (an average difference between extracts and residues of 78 ± 36 ‰), suggesting that non-extractable OC is older. Δ14C values of extracts and residues were strongly correlated and proportional to Δ14C values of bulk MOC but were not dependent on mineralogy. Neither MOC extractability nor differences in Δ14C values between extracts and residues changed with depth along soil profiles, where declining Δ14C values might indicate slower OC turnover in deeper soils. Thus, the 14C depth gradients in the studied soils were not explained by increasing stability of organic–mineral associations with soil depth.
Although H2O2 removed 90 ± 8 % of the MOC, the Δ14C values of oxidized OC (on average −50 ± 110 ‰) were similar to those of OC extracted with NaF–NaOH (−51 ± 122 ‰), but oxidation residues (−345 ± 227 ‰) were much more depleted in 14C than residues of the NaF–NaOH extraction (−130 ± 121 ‰). Accordingly, both chemical treatments removed OC from the same continuum, and oxidation residues were older than extraction residues because more OC was removed. In contrast to the NaF–NaOH extractions, higher contents of pedogenic oxides slightly increased the oxidation resistance of MOC, but this higher H2O2 resistance did not coincide with more negative Δ14C values of MOC nor its oxidation residues. Therefore, none of the applied chemical fractionation schemes were able to explain site-specific differences in Δ14C values. Our results indicate that total MOC was dominated by OC interactions with pedogenic oxides rather than clay minerals, as we detected no difference in bond strength between clay-rich and clay-poor sites. This suggests that site-specific differences in Δ14C values of bulk MOC and depth profiles are driven by the accumulation and exchange rates of OC at mineral surfaces.
- Article
(4814 KB) - Full-text XML
-
Supplement
(261 KB) - BibTeX
- EndNote





The persistence of organic matter (OM) in soil is a key control of atmospheric CO2 concentrations. Association of OM with minerals is considered an effective pathway of stabilizing otherwise degradable OM from microbial mineralization (Schmidt et al., 2011; Lehmann and Kleber, 2015; Hemingway et al., 2019), and, in many soils, the vast majority of organic carbon (OC) is bound to minerals (Kleber et al., 2015; Kögel-Knabner et al., 2008; Cotrufo et al., 2019). Despite the abundance of mineral-bound OC (MOC), we lack fundamental knowledge on the drivers of stability and turnover of MOC along soil profiles and across sites.
The formation of MOC involves sorption to reactive minerals such as phyllosilicate clays and pedogenic aluminium (Al) and iron (Fe) oxides, oxyhydroxides, and hydroxides (together referred to as oxides) (Schrumpf et al., 2013; Kögel-Knabner et al., 2008; Kaiser and Guggenberger, 2000; Khomo et al., 2017). Along with plant-derived decomposition products, microbial residues and metabolites are important precursors and sorbates for MOC formation (Avneri-Katz et al., 2017; Chenu and Stotzky, 2002; Cotrufo et al., 2015; Kalbitz et al., 2005; Kallenbach et al., 2016). Experimental studies have shown that sorption to minerals reduces OC mineralization (e.g. Kalbitz et al., 2005; Jones and Edwards, 1998; Eusterhues et al., 2014; Porras et al., 2018). Radiocarbon (14C) analyses confirmed the greater stability of MOC in soils, showing that on average the MOC age is older than OC not bound to minerals (Schrumpf et al., 2013; Kögel-Knabner et al., 2008; Hemingway et al., 2019; Heckman et al., 2018). Several field and incubation studies have suggested that the total amount and stability of MOC increases with the amount of pedogenic Al and Fe oxides (Bruun et al., 2010; Torn et al., 1997; Porras et al., 2017; Rasmussen et al., 2006). Other studies found correlations between Al and Fe oxides and MOC concentrations but not with the 14C content of MOC or the average age (Herold et al., 2014; Khomo et al., 2017; Schrumpf et al., 2013).
Studying MOC turnover and its drivers in soil is complicated because, similar to bulk soil OC, it is a mixture of younger and older carbon with a range of levels of stabilization (Trumbore et al., 1989; Swanston et al., 2005; Schrumpf and Kaiser, 2015; Koarashi et al., 2012). Various chemical methods, including acid hydrolysis and chemical oxidation, are commonly used to distinguish faster- and slower-cycling fractions (e.g. Mikutta et al., 2006; Jagadamma et al., 2010; Helfrich et al., 2007; Six et al., 2002; Paul et al., 2001; Eusterhues et al., 2003). All methods separate bulk soil OC or MOC into younger and older fractions but differ in the extent of OC removal and the 14C contents of the obtained residues. The oxidants H2O2 or Na2S2O8 were more effective in removing OC from samples than NaOCl, and their residues were older (Jagadamma et al., 2010; Helfrich et al., 2007). With the increasing presence of Al and Fe oxides in soils, larger amounts of OC resisted chemical oxidation, suggesting that binding to those minerals provides some protection against oxidative attack (Mikutta et al., 2006; Kleber et al., 2005; Eusterhues et al., 2005). However, there are indications that such chemically defined fractions are not causally related to MOC bioavailability or persistence in soils (Poirier et al., 2003; Mikutta and Kaiser, 2011; Helfrich et al., 2007; Jagadamma et al., 2010; Paul et al., 2008).
Release of mineral-bonded OC, either by desorption or upon mineral dissolution under changing environmental conditions, can support or even be prerequisite for its microbial degradation (Keil et al., 1994; Mikutta et al., 2007). Desorption of OM, although typically low under conditions similar to those during the formation of MOC (e.g. Gu et al., 1994), increases in the presence of competing ions such as SO or H2PO and is maximized by increasing the alkalinity of the solution (e.g. Kaiser and Zech, 1999; Kaiser and Guggenberger, 2007). While some studies observed an increased release of OC from minerals into alkaline solutions with greater mineral OC loading (Kaiser and Guggenberger, 2007; Kaiser et al., 2007), others showed increased OC desorption in subsoils despite lower OC concentrations (Kaiser and Zech, 1999; Mikutta et al., 2009). Greater desorption and biodegradation of OM bonded to phyllosilicates than of OM bonded to Al and Fe oxides has been attributed to differences in bonding strength. The sorption between OM and phyllosilicates is largely a result of the weaker cation bridges and van der Waals bonds, whereas the sorption of OM to Al and Fe oxides involves surface complexation, which results in strong chemical bonds (Singh et al., 2016; Mikutta et al., 2007). Thus, the presence and abundance of Al and Fe oxides typically decreased both desorption and mineralization rates (Oren and Chefetz, 2012; Saidy et al., 2012; Singh et al., 2017). Unfortunately, OC desorption has primarily been studied using model minerals in laboratory experiments, and observation times for desorption or mineralization were much shorter than carbon residence times in soil.
Mikutta et al. (2010) analysed the 14C contents of MOC after removing all potentially desorbable OC by extracting soil with a combination of NaF and NaOH. This extraction enables the study of the potential displacement of complexed organic functional groups by introducing OH− and F− anions to compete for and displace mineral-bound OC and by increasing the pH (Kaiser et al., 2007). Consistently younger OM was extracted from MOC of topsoils, supporting the idea that desorbable OC turns over faster than more strongly bonded OC. Results for subsoils were more variable (Mikutta et al., 2010). Along with 14C contents, the chemical composition of bulk soil OC extractable into alkaline solutions also changes with soil depth, with subsoils containing less lignin-derived aromatics but more O-alkyl C, possibly of microbial origin (Möller et al., 2000; Mikutta et al., 2009). Desorption of OC from MOC has, to our knowledge, not yet been studied systematically across soil types. If potential OC desorption better represents the mechanism of mineral protection of soil OC than the above-mentioned oxidative or hydrolytic extractions, then further research must focus on factors controlling the amount, age, and composition of desorbable OC in soils. Potential factors affecting MOC desorption include soil mineralogy, chemical composition of OM (and thus vegetation type), and OM loading on minerals.
Based on the literature review we expect the following:
-
Extraction in NaF–NaOH releases a weaker bound fraction of total MOC, which is younger than the stronger bound residue fraction.
-
The MOC of soil samples with higher proportions of extractable carbon is younger than MOC with more extraction-resistant OC.
-
The proportion of NaF–NaOH-extractable MOC decreases with increasing pedogenic oxide content, as pedogenic oxides form strong bonds with OC.
-
The chemical composition of extractable MOC varies between study sites due to differences in vegetation composition and, thus, litter chemistry.
-
The proportion of NaF–NaOH-extractable MOC declines with depth due to the declining OC loading of minerals.
-
The chemical composition of extractable MOC changes with depth due to the declining contributions of plant-derived OC and the increasing contributions of microbial-derived OC.
-
MOC should be less prone to desorption and, therefore, of older 14C age when organic molecules forming MOC have many carboxyl groups that enable strong bonds with minerals surfaces.
-
The strong oxidizing agent H2O2 removes more OC from MOC than NaF–NaOH.
-
Oxidizable and non-oxidizable OC should both be older than the extractable and non-extractable OC fractions if the harsher oxidation treatment removes more (and thus presumably better stabilized) OC from mineral surfaces, leaving OC residues of even older 14C age behind.
To test if maximum desorption with NaF–NaOH is a suitable indicator for the labile proportion of MOC, we took advantage of a previous experiment in which MOC was isolated from soils by density fractionation (heavy fraction, HF, at a density cut-off of > 1.6 g cm−3) for a range of sites across central Europe (Schrumpf et al., 2013). We selected five sites across ranges of clay content, type of clay and pedogenic oxides, and land use conditions, with associated differences in the amount and quality of litter input. Samples from three soil depths were extracted with a mixture of NaF and NaOH, and the extracted OC was analysed for amount, composition, and 14C content. We compared results from the extraction to the amount and age of OC oxidizable by heated H2O2, which was shown to remove the most MOC and isolate the oldest MOC (Helfrich et al., 2007; Jagadamma et al., 2010). The NaF–NaOH extraction experiment seeks to elucidate mineral protection as a stabilization mechanism, whereas the oxidation treatment explores the chemical recalcitrance of MOC.
The composition and age structure of MOC were studied on heavy fraction (HF) material obtained at five of the sites presented by Schrumpf et al. (2013). The sites include two deciduous forests developed on loess in Hesse (France, Luvisol, WRB, 2015) and loess over limestone in Hainich (Germany, Cambisol). Soils at the grassland site Laqueuille (France, Andosol) and the coniferous site Wetzstein (Germany, Podzol) are characterized by large pedogenic oxide contents. The soil at the fifth site, a cropland site in Gebesee (Germany, Chernozem), reveals a plow layer down to 30 cm and large contributions of old OC throughout the profile (Schrumpf et al., 2013). The MOC fraction was separated using two-step sequential density flotation in sodium polytungstate solution (1.6 g cm−3). After removal of the unprotected free light fraction in a first flotation step, samples were sonicated (site-specific energy application) to disrupt aggregates and separate the occluded light fraction from the targeted MOC in the HF (for details, see Schrumpf et al., 2013). Concentrations of OC in MOC are shown in Fig. S1 and ranged from 16.8 g kg−1 at Gebesee to 108 g kg−1 at Laqueuille in the uppermost layer, and from 4.6 g kg−1 at Hesse to 44 g kg−1 at Laqueuille in the deepest studied layer. Selected bulk soil properties of respective samples were adopted from Schrumpf et al. (2013) and are summarized in Table S1.
We randomly selected 3 out of the original 10 replicated soil cores per site and analysed the 0–5, 10–20, and 30–40 cm soil layers (excluding Wetzstein, where the 0–10, 10–30, and 30–50 cm layers were analysed from soil pits instead of cores; see Schrumpf et al., 2013) for (1) extractable MOC and (2) oxidation-resistant MOC.
NaF–NaOH-extractable MOC was determined as an indicator for maximal potential desorption by weighing 25 g of heavy fraction (MOC) material of each sample into 250 mL polypropylene centrifuge bottles and adding 125 mL of a 1:1 solution containing 0.8 M NaF and 0.2 M NaOH. Containers were then closed and agitated overnight (at least for 18 h) in an end-over shaker. A few drops of magnesium chloride were added as flocculant to the solution, which was then centrifuged for 15 min at 4000 × g. Then supernatants were decanted into 1000 mL polyethylene (PE) bottles and stored in the refrigerator. Another 125 mL of the extraction solution was added to settled soil material in the centrifuge tube, stirred, and mixed well to repeat the extraction a total of four times. Finally, the combined extract solution from each sample was passed through glass fibre filters with a pore size of 1.6 µm and stored in a 4 ∘C room until transfer into deionized water-rinsed, 75 cm long SERVAPOR 29 mm cellulose-acetate tubing for dialysis. The approximately two-thirds-full tubes were placed into clean 10 L buckets filled with deionized (DI) water, which was frequently replaced until the electrical conductivity of the external solution was < 2 µS. The content of the dialysis tubes was then freeze-dried and analysed for total C, N, and 14C signatures (as described below). The extracted residual soil containing the non-extractable OC was washed three times with deionized water to minimize remaining fluoride content before freeze-drying and analyses of total C and N contents as well as 14C signature (as described below).
Oxidation-resistant MOC was obtained following a slightly modified procedure from Jagadamma et al. (2010). After letting 2 g of soil soak in 20 mL of Millipore DI water for 10 min, 60 mL of 10 % hydrogen peroxide (H2O2) was gradually added to the soil. After the frothing had subsided from the reaction of wet samples with 60 mL of H2O2 at room temperature, the samples were heated and stirred regularly in a 50 ∘C water bath in order to catalyse the oxidation of organic matter. Because H2O2 decomposes with exposure to light and temperature, the samples were centrifuged, the supernatant decanted, and fresh H2O2 was added to continue the oxidation. Each sample was oxidized for two periods of 24 h and one period of 72 h. After the final oxidation, samples were centrifuged at 3500 × g for at least 15 min and then washed three times with 80 mL of deionized water. Magnesium chloride solution was added to enhance flocculation during centrifugation. Afterwards, samples were freeze-dried. Following this, the samples were homogenized using a ceramic ball mill and measured for total carbon and nitrogen content by dry combustion with the Vario EL CN analyser (Elementar Analysensysteme GmbH, Hanau, Germany). All soils analysed in this study were free of carbonate. Therefore, total carbon measurements are equivalent to total organic carbon in the soil.
Radiocarbon contents of the samples were measured on graphitized samples at the 14C laboratory in Jena, Germany (Steinhof et al., 2017). Soil samples were weighed into tin capsules and combusted in an elemental analyser. The evolved CO2 was transferred into a glass tube cooled by liquid nitrogen and then reduced to graphite at 600 ∘C under hydrogen gas atmosphere, using iron as the catalyst. The graphite was analysed by 14C accelerator mass spectrometry (AMS; 3MV Tandetron 4130 AMS 14C system; High Voltage Engineering Europa, HVEE, the Netherlands). Samples with low OC concentrations from the H2O2 residues were combusted with CuO wire in quartz tubes and graphitized using a sealed zinc reduction method; they were then analysed at the WM Keck Carbon Cycle AMS facility at UC Irvine (Xu et al., 2007).
Radiocarbon data are reported as Δ14C in per mille [‰], which is the relative difference in parts per thousand of the 14C 12C ratio of the sample with respect to an absolute standard (0.95 times the oxalic acid standard NBS SRM 4990C decay corrected to 1950), after normalization of the sample 14C 12C for mass dependent fractionation by normalizing to a δ13C of −25 ‰ (Trumbore et al., 2016). The average measurement precision of the Δ14C values was 2.8 ‰. Reported in this way, Δ14C values close to zero indicate that most of the C in the sample is close to that of the standard, i.e. it was fixed from the preindustrial atmosphere (up to ∼ 350 years prior to 1950). Negative Δ14C values indicate lower 14C 12C than the standard and specify that most of the C in the sample had been isolated from the atmosphere long enough for detectable radioactive decay to deplete 14C; samples with negative Δ14C values have been isolated longer than 400 years. Positive Δ14C values indicate enrichment with bomb-derived 14C from nuclear weapon testing in the early 1960s, which suggests that OC from these samples is younger and mostly fixed in the last century. In 2004, when the samples in this study were collected, atmospheric Δ14C values (about 70 ‰; Levin et al., 2010) were still declining from the peak bomb C in 1963 of about +900 ‰. Thus, comparing oxidized or extracted C with the original mineral-associated OM (MOM) depends on its original Δ14C values. For total MOM with negative Δ14C values, we assume the extracted or oxidized C is “younger” if it has Δ14C values that are either less negative or positive. For total MOM that initially has positive Δ14C values, we might expect “older” C residues to have either higher Δ14C values (closer to the bomb peak) or negative Δ14C values; in both cases, we expect “younger” extracted Δ14C values to have positive Δ14C values. Our previous results, based on temporal changes in Δ14C values of MOC at the Hainich site (Schrumpf and Kaiser, 2015), indicate that total MOC in our soils will have turnover times of > 20 years. Therefore, if extracted or oxidized OC has Δ14C values that are higher than unextracted or residual MOM, we refer to these as younger throughout the paper.
To determine the amount of carbon in the residues from both extraction procedures, we multiplied the measured OC content in the recovered residues by its mass. The amount of carbon lost by the treatments was determined as the difference between the original OC content of the HF sample and the OC in the residues. The radiocarbon contents of the H2O2 residues were directly measured, and the 14C fraction of the OC lost or extracted (14Cextract) was determined by mass balance as follows:
The same formula was used to determine the 14C fraction of OC extracted by NaF–NaOH. As OC extracted by NaF–NaOH extraction was also measured directly, the mass balance results allow for the identification of potential bias in the measured 14C data caused by losses of extracted OC during dialyses.
Solid-state cross-polarization magic angle spinning 13C-nuclear magnetic resonance (CPMAS-13C-NMR) spectra of NaF–NaOH-extracted, dialysed, and freeze-dried OM were recorded on an AVANCE III spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) at a resonance frequency of 100.5 MHz, with a proton spin-lock and decoupling frequency of 400 MHz. The proton nutation frequency was 80 kHz, corresponding to a pulse duration of 3.12 µs. The cross-polarization time was 500 µs. Samples were weighed into 4 mm zirconium oxide rotors that were spun at 10 kHz around an axis declined by the “magic angle” of 54.74∘ against the static magnetic field; the contact time was 1 ms, and the recycle delay time was set to 0.4 s. Depending on the sample, between 4000 and 35 000 scans were recorded. The spectra were processed with a line broadening of 100 Hz. Chemical shifts are given relative to the resonance of tetramethylsilane. After baseline correction, the intensities of spectral regions were corrected for different cross-polarization efficiencies in different spectral regions. To do so, the 1H spin-lattice relaxation time in the rotating frame (T1ρ) as well as the cross-polarization build-up time for the magnetization transfer from 1H to 13C (TCH) were estimated selectively for the regions. As OM samples with C contents > 30 % were analysed, no treatment for the removal of paramagnetic mineral phases was required, and the obtained spectra were well resolved and showed no indications of paramagnetic interferences. In addition, we analysed one bulk MOC sample that was low in pedogenic oxides without the removal of paramagnetic phases. In all cases, spectra were reasonably well resolved and showed no indications of paramagnetic interferences.
Resonance areas were calculated by electronic integration: alkyl region (0–50 ppm), mainly representing C atoms bonded to other C atoms (methyl, methylene, and methine groups); O/N-alkyl region (50–110 ppm), mainly representing C bonded to O and N (carbohydrates, alcohols, and ethers) and including the methoxyl C (peak centred around 56 ppm); phenolic and aromatic region (110–160 ppm), representing C in aromatic systems and olefins; and the carbonyl region (160–220 ppm), including carboxyl C (160–190 ppm). Further information on the assignment of 13C-NMR regions are given by Wilson (1987) and Orem and Hatcher (1987).

Figure 1Proportion of OC extractable with NaF–NaOH or oxidized with heated H2O2 from the mineral-associated OC fractions of soil samples from five different sites (Hainich, Hesse, Laqueuille, Wetzstein, and Gebesee) and three soil depths (0–5, 10–20, and 30–40 cm); for Wetzstein the studied soil depths were 0–10, 10–30, and 30–50 cm. “*” denotes that different soil depths were studied at the Wetzstein site.
3.1 Mineral-associated organic carbon extracted by NaF–NaOH
The NaF–NaOH extraction removed on average 58 ± 11 % of bulk MOC across sites and soil depths (Fig. 1). The extraction efficiency was not explained by soil depth, as the average extraction removal values were 57 ± 7 % in the uppermost, 60 ± 15 % in the intermediate, and 56 ± 11 % in the deepest analysed soil layers. The average extraction efficiency was lower at the Gebesee site compared with other sites (41 ± 10 % versus 62 ± 11 % on average across depths). The amounts of extracted and residual OC, which represent similar proportions of bulk MOC, were strongly correlated with bulk MOC concentrations across sites and soil depths (r=0.96, p<0.01; Figs. 2, S2). The pedogenic oxide contents, the OC loadings of minerals, and the soil pH did not affect the proportion of extractable MOC. However, as the bulk MOC concentration was positively related to the pedogenic oxide contents (Schrumpf et al., 2013), the same relationship applied to OC concentrations in extraction residues (Fig. 3).
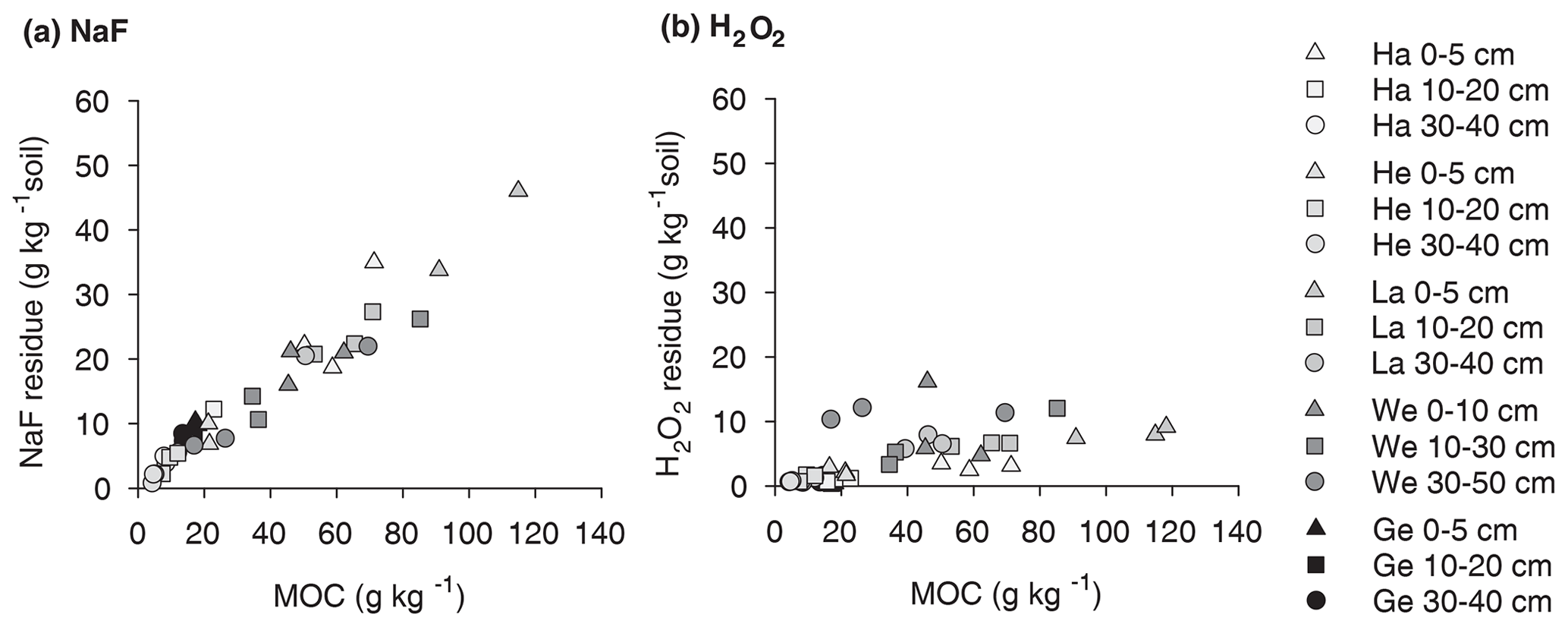
Figure 2Residual OC depended on original concentrations of mineral-associated OC for (a) NaF–NaOH extraction and (b) H2O2 oxidation for Hainich (Ha), Hesse (He), Laqueuille (La), Wetzstein (We), and Gebesee (Ge).
3.2 Radiocarbon contents of NaF–NaOH extracts and residues
Directly measured Δ14C values of dialysed NaF–NaOH extracts were comparable to calculated Δ14C values in extracts using the mass balance approach (Fig. 4), suggesting that there were no systematic losses of older or younger C during the extraction and subsequent dialysis procedure. The only exceptions were the results from Gebesee, where the mass balance approach suggests that some young carbon was lost during dialysis of the extracts.
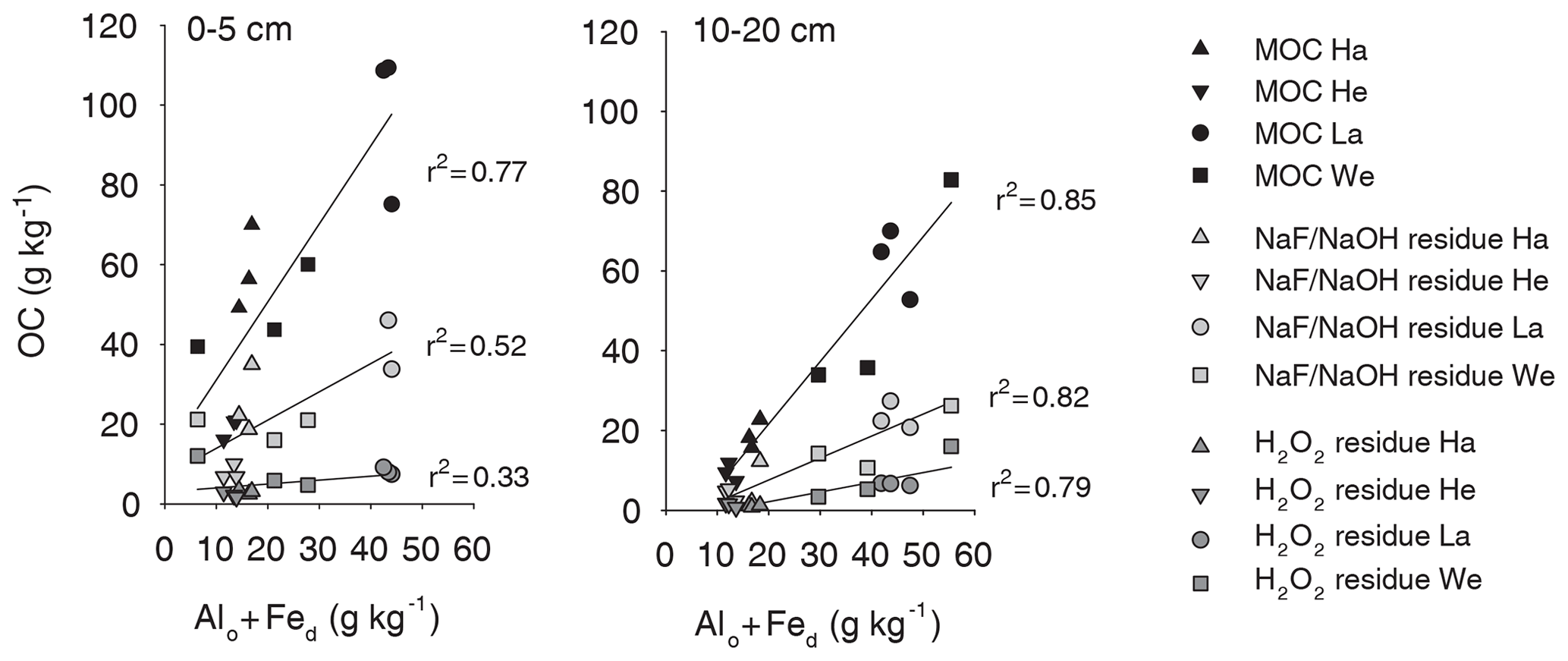
Figure 3The OC concentrations in each fraction (bulk MOC, NaF–NaOH extraction residue, and oxidation residue) were related to the pedogenic oxide content of the sample, which is expressed as the sum of the acid oxalate-extractable Alo and dithionite-extractable Fed.
Extracted fractions had consistently higher Δ14C values than extraction residues, and Δ14C values decreased with depth at all sites (Fig. 5). The Δ14C values of the OC extracted from the uppermost layers increased from −126 ‰ to 142 ‰ in the following order: Gebesee (Chernozem) < Wetzstein (0–10 cm, Podzol) < Laqueuille (Andosol) < Hainich (Cambisol) < Hesse (Luvisol). The average difference in Δ14C between extracted and residue OC was 79 ± 36 ‰ across sites and increased in the following order: Gebesee (34 ± 4 ‰) < Laqueuille (38 ± 6 ‰) < Hainich (63 ± 3 ‰) < Hesse (84 ± 5 ‰) < Wetzstein (100 ± 15 ‰). As indicated by the almost parallel shifts in Δ14C values, there was no general trend for increasing or declining differences in 14C contents with soil depth (Fig. 5). Instead, Δ14C values of extracts and extraction residues were highly correlated (r2=0.91, p<0.01, Fig. S3). However, Δ14C values of bulk MOC and extractable or residual MOC were all unrelated to total MOC or its extractability (results not shown).

Figure 4Comparison of 14C contents of NaF–NaOH extracted OC obtained using a mass balance approach with direct measurements of the extracts after dialyses for (a) Hainich (Ha), Hesse (He), Laqueuille (La), and Wetzstein (We), and (b) Gebesee (Ge).
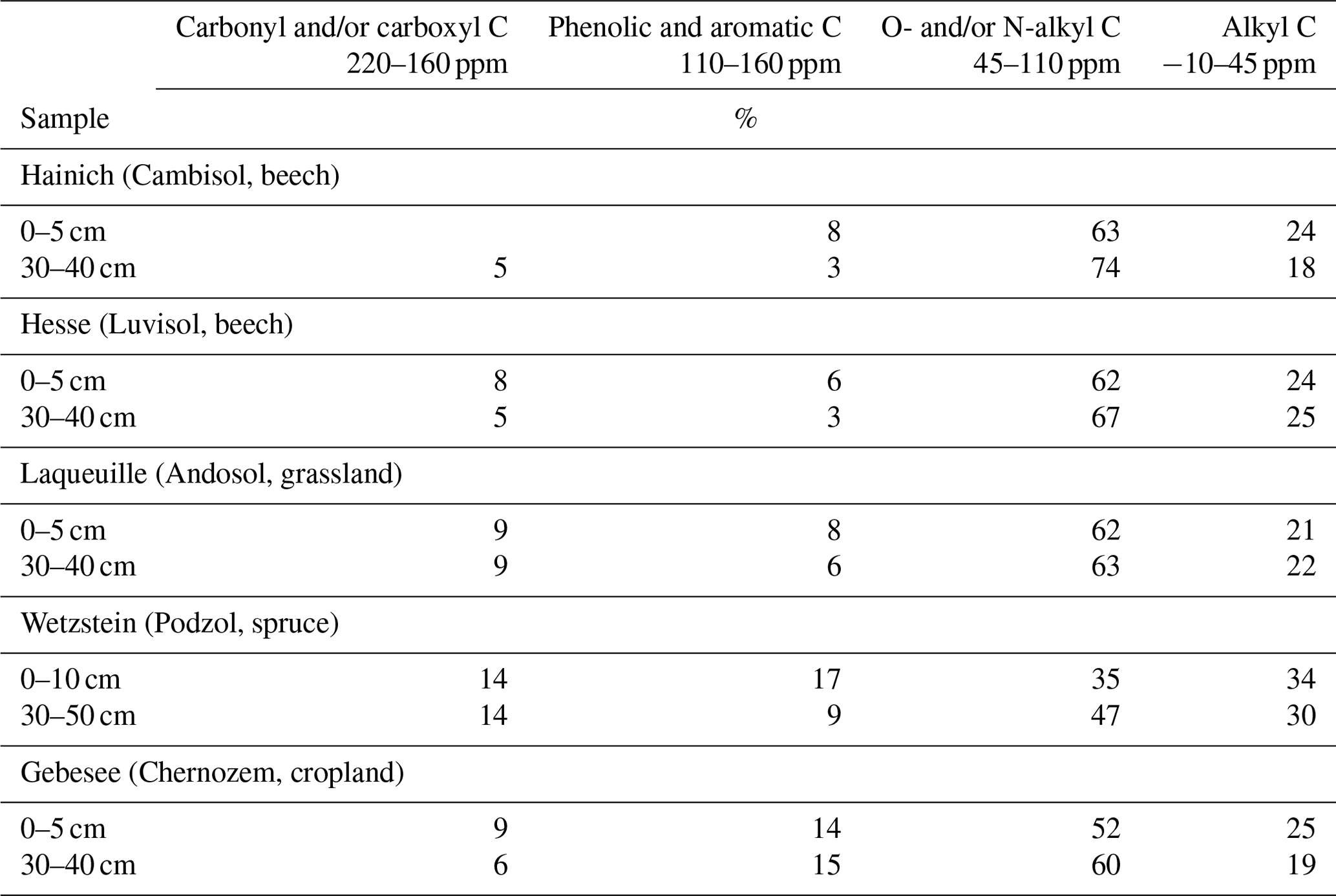
Table 1Distribution of C species in organic matter extracted into NaF–NaOH from heavy fractions of mineral topsoil (0–5 cm depth) and subsoil (30–40 or 30–50 cm depth) layers as revealed by CPMAS-13C-NMR.
3.3 NMR spectroscopy
The NMR spectra of extracted OM from soils of the Hainich, Hesse, and Laqueuille sites were remarkably similar and were dominated by signals in the O/N-alkyl C region (62 %–74 % of total peak area across sites and depths) and the alkyl C region (18 %–25 %), suggesting a strong contribution of carbohydrates and aliphatic compounds to the extracted OM (Fig. 6, Table 1). All six spectra also reveal a distinct peak centred around 174 ppm, due to carboxyl C, which is in line with the fact that the alkaline extraction tends to preferentially release acidic compounds (5 %–9 %). All spectra show small signals in the aromatic regions centred around 150 %ppm (phenols) and 130 %ppm (non-substituted aromatic systems). All six spectra featured signals, some even well-resolved, around 56 %ppm, indicating the presence of methoxyl C.
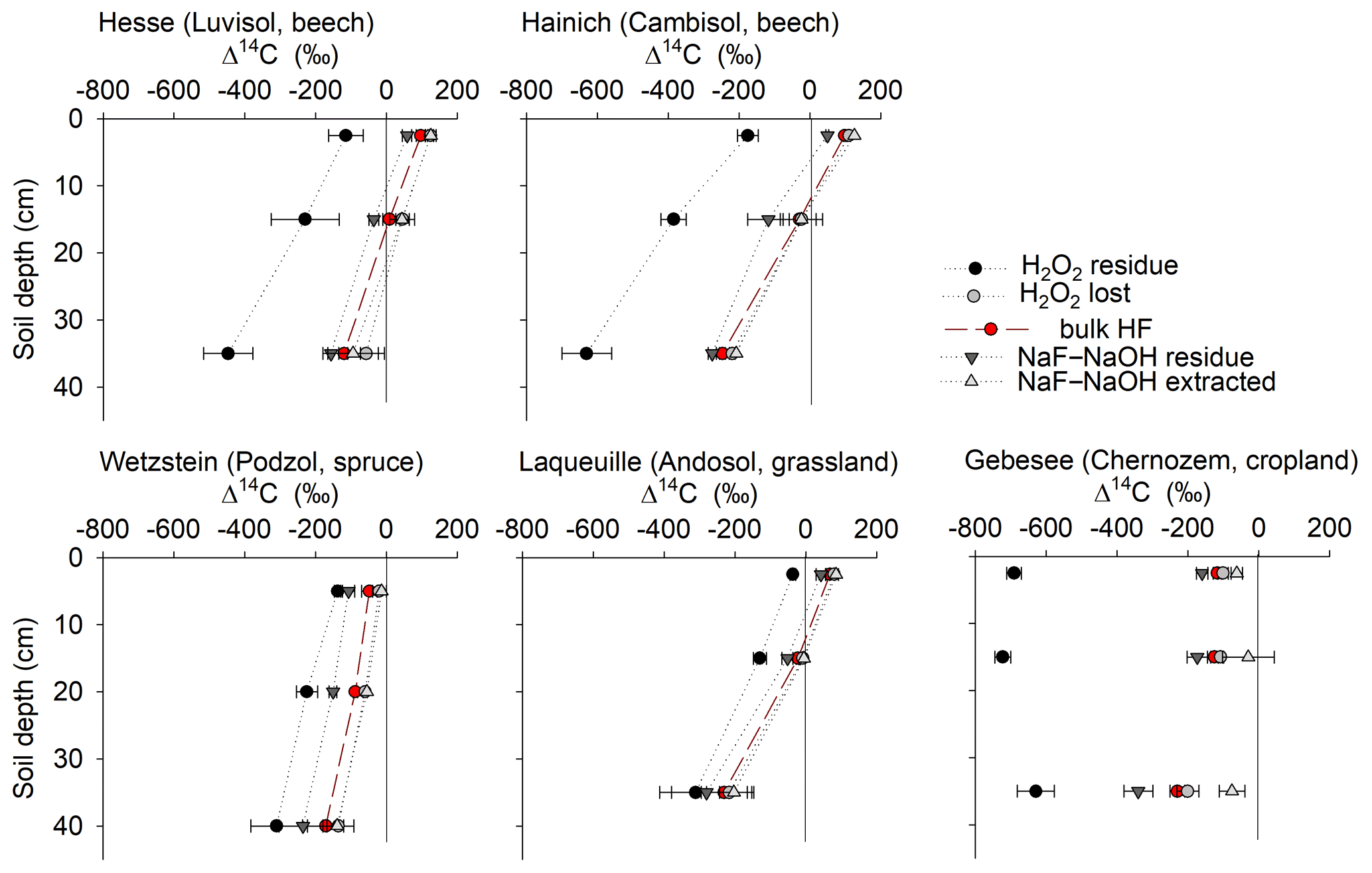
Figure 5Depth profiles of radiocarbon (Δ14C) in bulk mineral-associated OC (bulk MOC), as well as in OC removed from mineral surfaces using either NaF–NaOH or H2O2, and in the respective OC residues remaining on mineral surfaces.
The spectra obtained on OM extracted from the Chernozem-type soil at site Gebesee resembles those of the Hainich, Hesse, and Laqueuille sites, except for the fact that they indicate more non-substituted aromatic systems, which is consistent with the occurrence of pyrogenic OM in this soil type. When comparing the spectra of OM from top- and subsoils, differences were surprisingly small at Hainich, Hesse, Laqueuille, and Gebesee, with proportions of alkyl and aromatic C tending to decrease in favour of increased signals of O/N-alkyl C. The contribution of carbonyl/carboxyl (carbonyl and/or carboxyl) C remained constant with soil depth.
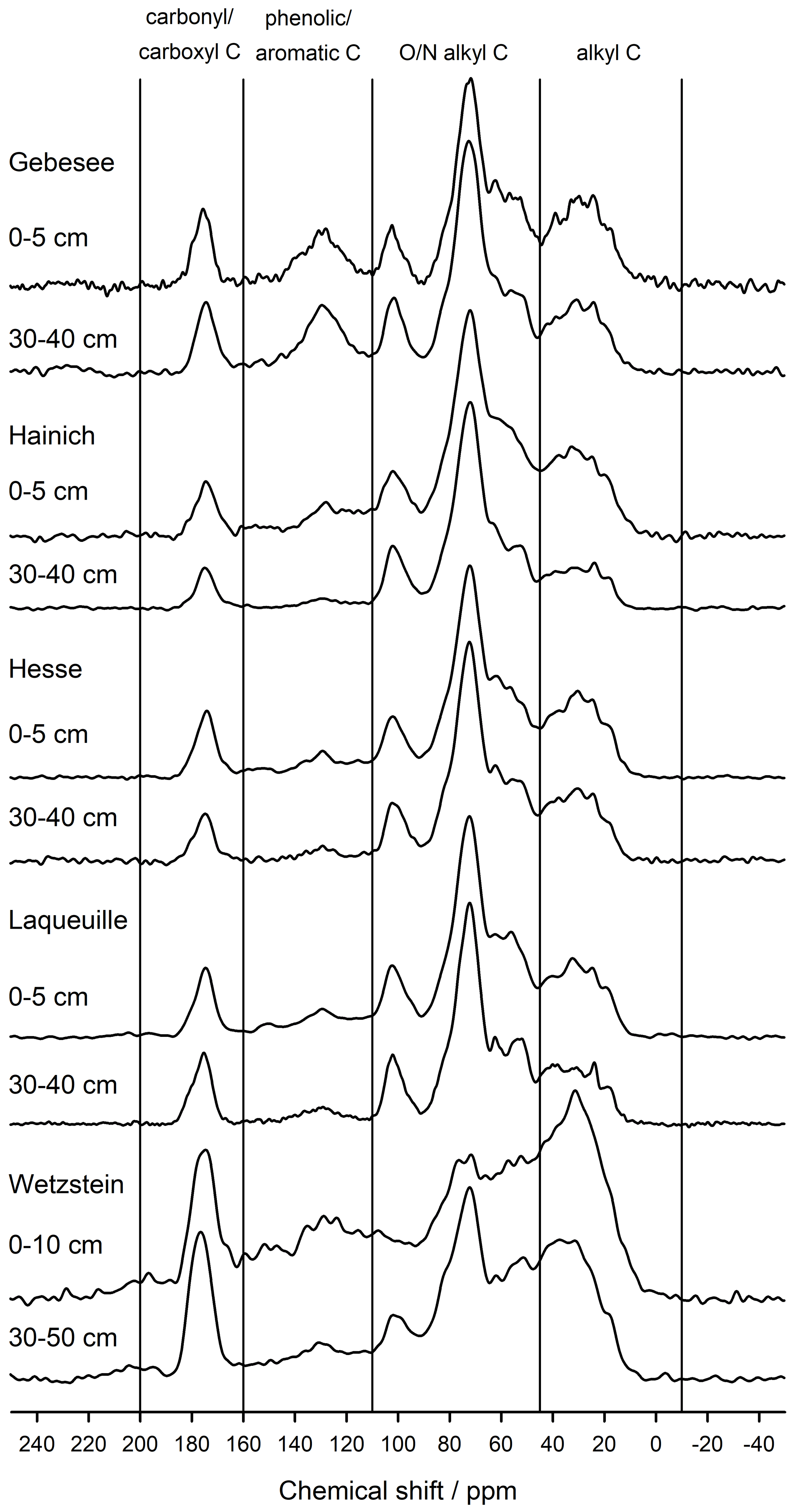
Figure 6CPMAS-13C-NMR spectra of OM extracted into NaF–NaOH from the mineral-associated fraction of two soil depths from the five study sites: Gebesee (Chernozem, cropland), Hainich (Cambisol, beech), Hesse (Luvisol, beech), Laqueuille (Andosol, grassland), and Wetzstein (Podzol, spruce).
The spectra obtained on extracted OM from the Podzol-type soil at Wetzstein showed the strongest deviation in spectral features. The signals due to carbonyl/carboxyl, phenolic and aromatic, and alkyl C were much more prominent than in all other spectra. Also, the change in composition with depth was much more evident for the Podzol site Wetzstein than for the others. Here, phenolic and aromatic as well as alkyl C decreased whereas O/N-alkyl increased, reflecting the strong redistribution of OM along the profile during podzolization. The composition of the subsoil OM at Wetzstein approached that of the OM at the Hainich, Hesse, and Laqueuille sites.
To understand how the extract differed from the bulk and residual fractions, we also analysed the bulk MOM and the NaF–NaOH extraction residue of the Hainich 0–5 cm sample. The signal-to-noise ratio of these spectra was lower than in the extracted OM due to the presence of paramagnetic minerals (Fig. 7). Nevertheless, the results reveal larger proportions of carbonyl/carboxyl C and especially phenolic and aromatic C but less O/N-alkyl C for the extraction residue than in the extracted OM (Fig. 7, Table 2).
Table 2Distribution of C species in total, NaF–NaOH-extractable, and residual organic matter of the heavy fraction of the mineral topsoil layer (0–5 cm depth) at Hainich (Cambisol, beech) as revealed by CPMAS-13C-NMR.
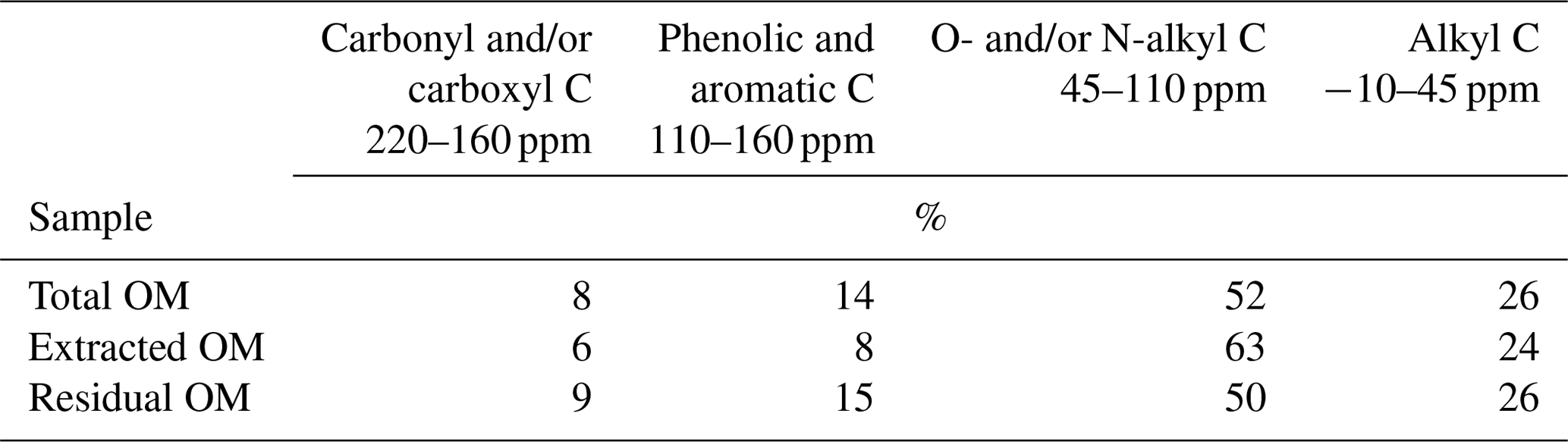

Figure 7CPMAS-13C-NMR spectra of OM from the 0–5 cm layer of the Hainich site (Cambisol, beech). (a) Total mineral-associated OM (MOM); (b) MOM fraction extracted into NaF–NaOH; (c) residual OM after extraction of MOM into NaF–NaOH.
3.4 MOC oxidation in heated H2O2
On average, only 11 ± 6 % of MOC resisted treatment with heated H2O2, with 90 ± 8 % of MOC oxidized in the uppermost layer, 90 ± 6 % oxidized in the intermediate layer, and 87 ± 5 % oxidized in deepest studied soil layer (Fig. 1). The proportion of MOC resisting the H2O2 treatment differed between study sites. The largest proportion of H2O2-resistant MOC was found at the Wetzstein site (Podzol, on average 19 ± 4 %), especially in the deepest layer, whereas the lowest proportions of H2O2-resistant MOC occurred at Gebesee (Chernozem, 5 ± 3 %). The absolute amount of H2O2-resistant OC was correlated with the bulk MOC concentration (r=0.76, p<0.01; Fig. 2), but the correlation was weaker than the one found for residual OC after NaF–NaOH extraction and bulk MOC.
Residues of the H2O2 treatment had much lower Δ14C values than residues of the NaF–NaOH extractions (Fig. 5). The average Δ14C values of H2O2 residues ranged between −36 ± 8 ‰ (Laqueuille) and −691 ± 21 ‰ (Gebesee) in the uppermost layer and between −310 ± 73 ‰ (Wetzstein) and −630 ± 70 ‰ (Hainich) in the deepest layer (Fig. 5). The difference between Δ14C values of oxidized and residual OC in the uppermost layer increased in the following order: Laqueuille = Wetzstein (115 ‰) < Hesse (239 ‰) < Hainich (290 ‰) < Gebesee (591 ‰); and they increased slightly with soil depth at Hesse, Hainich, and Wetzstein.
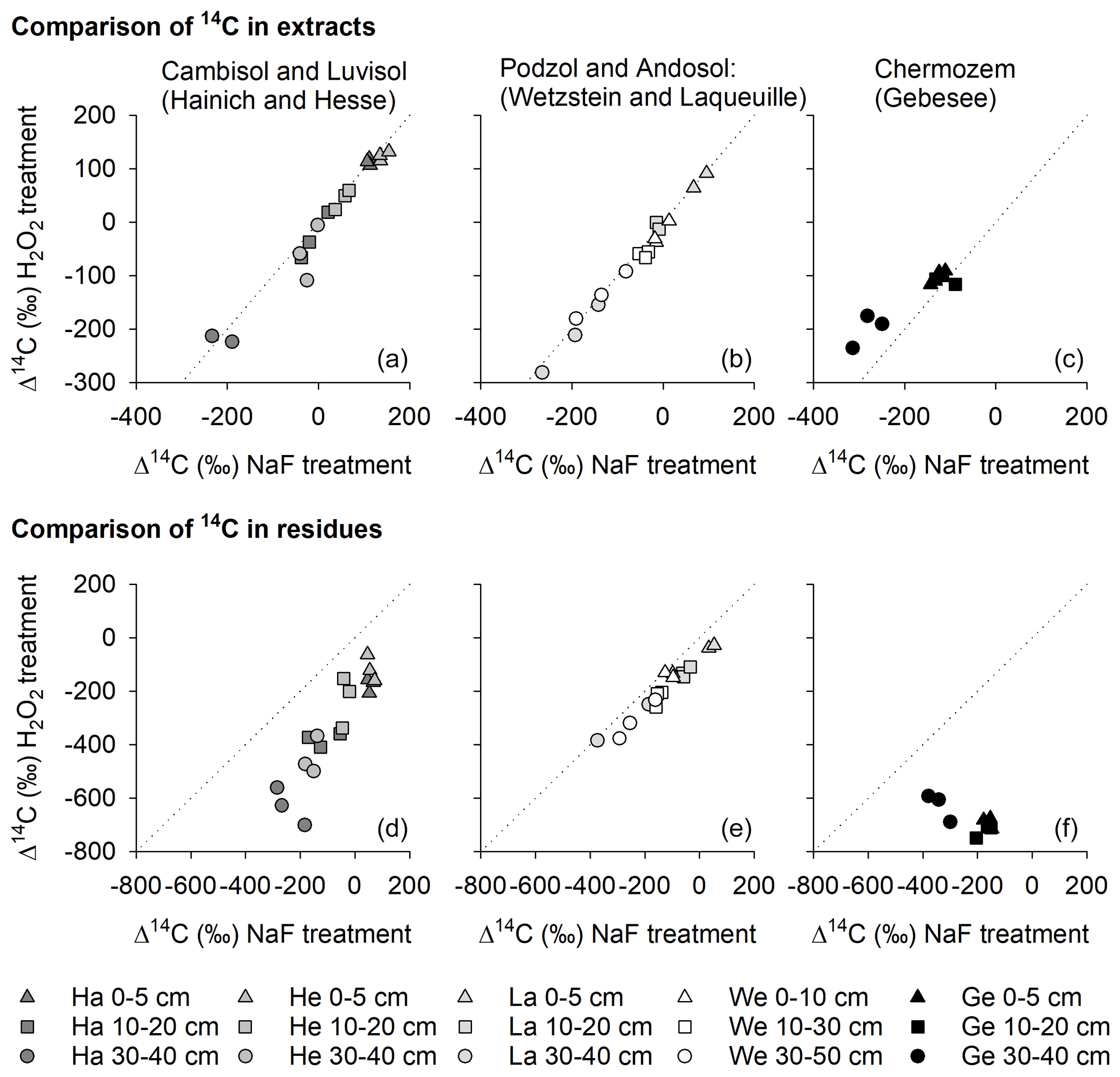
Figure 8Relationships between the radiocarbon content of OC removed from mineral surfaces by H2O2 and NaF–NaOH (a–c) and the radiocarbon content of OC residues after treatment with H2O2 and NaF–NaOH (d–f) for the Hainich (Ha), Hesse (He), Laqueuille (La), Wetzstein (We), and Gebesee (Ge) study sites.
When comparing Δ14C values of OC removed by either NaF–NaOH or H2O2 treatments, we were surprised to find no 14C difference for most sites (Fig. 8), despite different amounts of OC being removed by the individual procedures. The almost parallel shift in Δ14C values of H2O2 residues from NaF–NaOH residues in all soil profiles indicate that both were highly correlated within depth profiles (Fig. 8). For the sites rich in pedogenic oxides, Wetzstein and Laqueuille (Podzol and Andosol), Δ14C values in NaF–NaOH residues deviated from H2O2 residues by only 62 ± 26 ‰. In contrast, the difference in extraction and oxidation residual Δ14C values were 258 ± 99 ‰ for the Luvisol and Cambisol sites Hesse and Hainich (Fig. 8). The only exception was again the Chernozem site (Gebesee), where OC extracted by NaF–NaOH was on average younger than OC removed by H2O2, and the 14C contents of the two residues differed on average by 456 ± 135 ‰ and were not correlated.
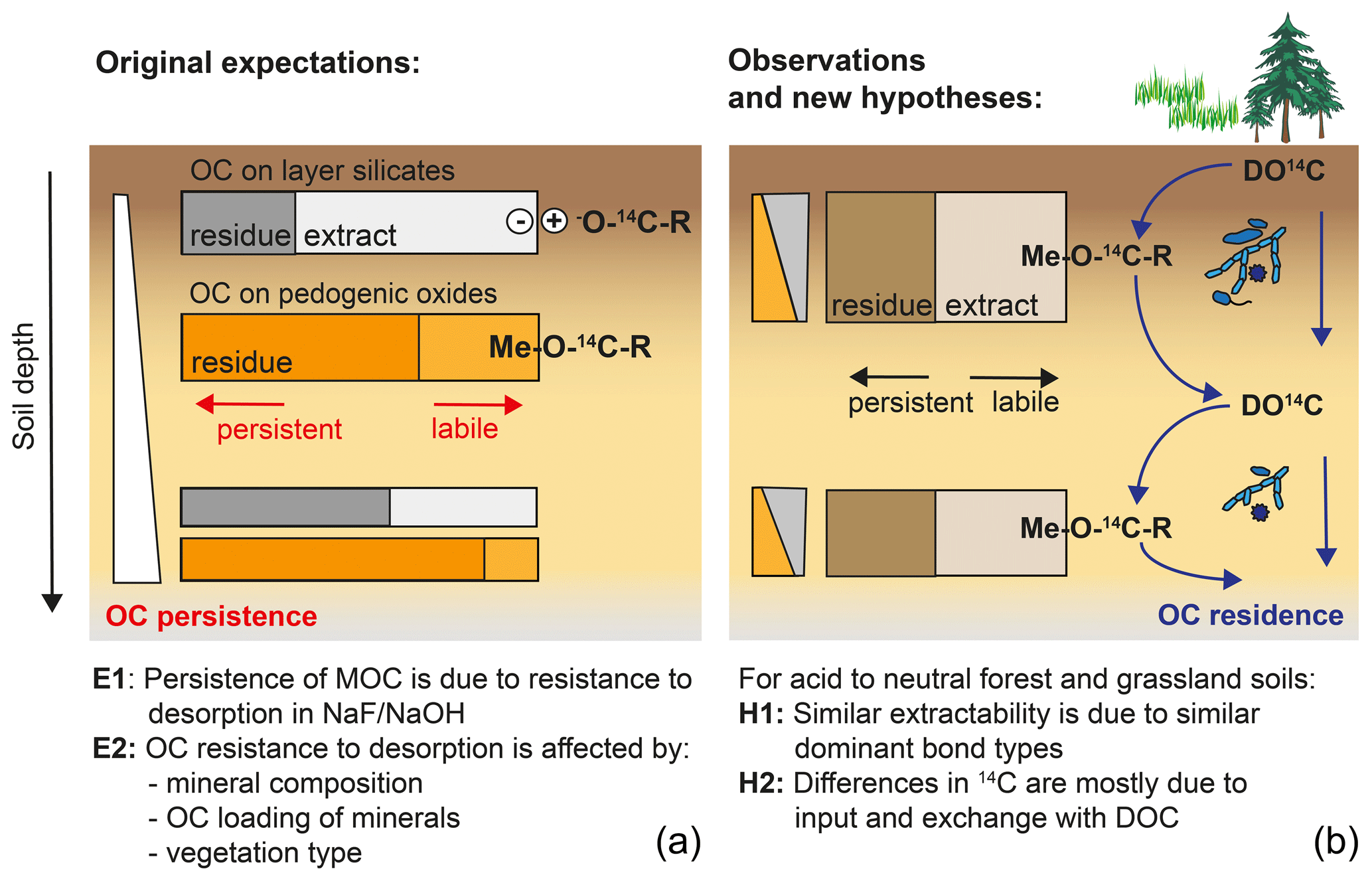
Figure 9Graphical summary of (a) original expectations where potential MOC desorption explains MOC persistence and varies with mineral composition and soil depth, and (b) the observation of site-independent desorption and alternative hypotheses explaining variations in Δ14C values of MOC across sites and with depth.
4.1 Unexpected similarity of the NaF–NaOH-extractable MOC
Strong hysteresis, rendering part of adsorbed OC resistant to desorption, is a common phenomenon found in sorption-desorption experiments with OM (Gu et al., 1994; Oren and Chefetz, 2012). Given that desorption rates in ambient soil solutions are low, we consider NaF–NaOH extractability an indicator for maximum potential desorption. Because the NaF–NaOH extraction offers ideal desorption conditions of the combination of competing OH− and F− anions and alkaline conditions, the method targets OC bound to minerals by Coulombic forces and surface complexation. The OC desorbed likely also includes OC held by different weaker forces, such as hydrogen bonds, cation bridges, or hydrophobic interactions.
We expected that the proportion of extractable C would increase with OC loading of minerals and, thus, be higher in topsoils with larger MOC concentrations than in subsoil layers with smaller concentrations. As minerals have different characteristic dominant binding modes for OC at a given pH (Mikutta et al., 2007), we further assumed that extractability would depend on mineral composition and soil pH, with the lowest desorption in acidic soils with larger amounts of pedogenic oxides (i.e. the Podzol or the Andosol site in our study). Finally, we expected that land-use- and site-specific differences in OM quality would influence MOC extraction. Our results showed, however, that a surprisingly constant proportion of on average 62 ± 11 % of the MOC was extracted by NaF–NaOH for our four uncultivated test sites, irrespective of soil depth, study site, or original OC concentration. Thus, despite the presumed variation in the chemical composition of litter input and the mineral assemblage between sites, actual interactions between soil OC and minerals were uniform.
Laboratory experiments showed similar proportions of extracted OC by NaF–NaOH from two Fe oxides, despite differences in the absolute amounts of OC sorbed by goethite and ferrihydrite (Kaiser et al., 2007). The amounts extracted from these model MOC experiments were surprisingly similar to the results of this study (around 65 %, Kaiser et al., 2007). Results from another experiment in which OC was extracted with NaOH from experimentally produced MOC on goethite also extracted a similar fraction of MOC (57 %–60 %, Kaiser and Guggenberger, 2007). We expected to observe more extractable MOC at sites poor in Al and Fe oxides but rich in clay minerals (such as the Cambisol at Hainich with 50 %–70 % clay), which we expected would have a larger share of weakly bound OC on MOC. It is possible that some of the weakly bonded OC was already lost in the preceding density fractionation with Na polytungstate solution (Schrumpf et al., 2013). However, the uniform extractability suggests that the extracted OC was bound to minerals by the same mechanism, irrespective of mineral composition. This indicates that either the same dominant bond mechanism operates for different minerals or that extracted OC originated predominantly from one mineral type. While OC adsorbed by covalent bonds is usually associated with pedogenic oxides, covalent bonds have also been shown to bind OC on the edges of clay minerals. (e.g. Chen et al., 2017; Gu et al., 1994). In our study, the linear relation between MOC and extraction residues with the sum of oxalate-extractable Al and dithionite-extractable Fe indicates that pedogenic Al and Fe oxides were important for OC binding across the study sites (Figs. 3, S3). Even very high clay contents (> 50 %), such as at the Hainich site, could not compensate for lower pedogenic oxide content, resulting in lower MOC storage. Therefore, the measured uniform extractability of MOC is likely an immediate result of the pedogenic oxides controlling MOC accumulation.
The similar chemical composition of MOC extracted from the Hainich, Hesse, and Laqueuille sites further supports a uniform mechanism dominating the association of organic C with minerals, especially given the lack of variation with pedogenic oxide content. One explanation could be that the type of sorptive interaction targeted by the NaF–NaOH extraction selects for a specific composition of the extracted MOC. Alternatively, the similar MOC composition may reflect uniform microbial processing of the organic input (Liang et al., 2017). However, the different chemical composition at the Gebesee and Wetzstein sites suggest that site-specific properties and pedogenic properties can indeed influence MOC composition. For the Chernozem at Gebesee sites, higher contents of non-substituted aromatic systems are likely due to a different vegetation history, where fire also played a role. Differences in OM composition between the Wetzstein and Laqueuille sites were surprising, as the pedo-environmental conditions (acidity and mineralogy) of OM accumulation in Podzol-type and Andosol-type soil are often considered to be similar (Aran et al., 2001; Young et al., 1980). The special MOC composition in the podzol-type soil is likely due to the special decomposition conditions at that site (conifer-dominated vegetation, pH, microbial community, and soil climate), which subsequently uniquely influence the dissolved organic carbon (DOC) production and chemistry at that site.
Experiments on model minerals suggested that desorption increases with increasing OC loading of minerals, i.e. when more OC is bound per mineral content of a sample (Kaiser and Guggenberger, 2007). This is consistent with our observation of reduced NaF–NaOH extraction at the agricultural soil site of Gebesee, where OC loading of minerals is reduced relative to undisturbed soils due to lower inputs, increased mineralization, and soil mixing by plowing (Plante et al., 2005; Helfrich et al., 2007). Although Fig. 3 shows that for the same amount of pedogenic oxides, less OC was bound to MOM in subsoils, soil depth did not affect MOC extractability in this study. This result is supported by studies by Mikutta et al. (2010) and Möller et al. (2000), which observed no significant decrease in NaF–NaOH extractability of OC with soil depth. One explanation for the uniform extraction with soil depth could be that DOC input to subsoils occurs mostly along specific flow paths (Bundt et al., 2001), enabling exposed mineral surfaces in subsoils to have similar OC loads as topsoils. Additionally, increasing pH with soil depth could reduce the sorption capacity of minerals in subsoils. Assuming that pH reduces the total number of available sorption sites at depth, the same proportion of available sorption sites could be occupied in subsoils as in topsoils, despite a smaller ratio of OC-to-pedogenic oxide contents. Whatever the mechanism, our findings confirm that a decline in potential desorption is not responsible for greater subsoil OC stability (Rumpel and Kögel-Knabner, 2011) and cannot explain the increased carbon ages of MOC with depth.
4.2 MOC desorption and Δ14C values
Results of 14C analyses of NaF–NaOH extracts and residues confirmed our hypothesis regarding the preferential extraction of younger carbon, with lower Δ14C values in residues compared with extracts or unextracted MOM across soil types and depths. These findings indicate that desorption facilitates the exchange of old OC for new OC on mineral surfaces, whereas non-extractable OC is less frequently exchanged. Using Na pyrophosphate as the extractant, Heckman et al. (2018) also observed consistently younger OC in MOC extracts and older OC in extraction residues across different soils and soil depths. We expect that Na pyrophosphate would have a similar effect on MOC as NaF–NaOH due to the comparable increase in pH and because both pyrophosphate and fluoride act as chelating agents to strongly compete with OC for binding sites on mineral surfaces.
Some MOC may be resistant to extraction due to multiple chemical bonds binding the OC with greater strength to mineral surfaces (Kaiser and Guggenberger, 2007). As multiple bonds require a higher number of involved functional groups, OC of extraction residues of multiple bonded OC should be enriched in carboxylic groups. Our NMR data support the resistance mechanism of multiple bonds, showing a higher share of carbonyl/carboxyl groups in non-extractable than in extractable MOC from the Hainich site. Phenolic and aromatic groups, which are also known to bind preferentially and strongly to mineral surfaces (Chorover and Amistadi, 2001; Kothawala et al., 2012; Avneri-Katz et al., 2017), were also enriched in the residues, whereas O/N-alkyl C was depleted. Given this mechanism, we expect the non-desorbable OC to be composed of aromatic and other compounds strongly bound to mineral surfaces by multiple functional groups, but this remains to be tested in future studies.
Our results did not confirm our original expectation that the average age of MOC is inversely related to its extraction in NaF–NaOH, due to the strength of the stabilizing chemical bonds in non-desorbable MOC. This may be a direct consequence of the small variation in OC extractability between samples, and it shows that extraction in NaF–NaOH is not able to explain between-site variation in Δ14C values.
4.3 Comparison between NaF–NaOH extraction and H2O2 oxidation results
As expected, residues of H2O2 treatments were older than residues of NaF–NaOH extractions. We further hypothesized that the heated H2O2 treatment would remove a larger and on average more stable fraction; therefore, the oxidized OC would have an older mean age than the extracted OC. While H2O2 removed on average 89 % of bulk HF OC and NaF–NaOH removed only 62 %, the 14C ages of MOC removed by oxidation were surprisingly comparable to those extracted by NaF–NaOH. This result indicates that NaF–NaOH residues contain oxidizable OC of a similar or only slightly older age than the extracted material. We found that both NaF–NaOH and H2O2 mostly removed OC from a younger, 14C-enriched pool, resulting in increasingly old residual OC as more OC is removed. Jagadamma et al. (2010) similarly observed that sequentially stronger oxidants removed C with similar 14C contents, irrespective of the extent of OC removal. Applying a mass balance approach to the results of the extraction procedures used by Helfrich et al. (2007) showed similar soil 14C contents of removed OC, irrespective of the extracted OC amounts. Accordingly, a large proportion of MOC is more homogenous in 14C contents than previously thought, whereas only a small proportion is very old.
The Δ14C difference between NaF–NaOH and oxidation residues was unexpectedly smaller for the two soils rich in pedogenic oxides (Laqueuille and Wetzstein) compared with the Luvisol and the Cambisol. The soils rich in pedogenic oxides had slightly higher amounts of OC left in oxidation residues, suggesting that they protected a larger proportion of OC against oxidation. Eusterhues et al. (2005) similarly observed that more OC resisted H2O2 oxidation in subsoils rich in pedogenic oxides and in older residues in the Dystric Cambisol than the Haplic Podzol studied. This study, in combination with our results, suggests that high contents of pedogenic oxides in soils increase the oxidation resistance of MOC but do not increase residue ages. Accordingly, oxidation is also likely not the dominant mechanism for exchange of OC on these mineral surfaces. The relatively young oxidation residues at Wetzstein and Laqueuille in our study could be due to high DOC fluxes and, thus, overall faster OC replacement of all MOC components at the Podzol site Wetzstein as well as the younger soil age of the Andosol soil at Laqueuille. At some of the sites, it is also possible that old oxidation-resistant OC was inherited from the parent material (e.g. the loess layer or limestone residues at Hainich and Hesse) or a specific fire history (e.g. at the Chernozem site Gebesee, where only small, but very old amounts of OC were left after the H2O2 treatment).
4.4 Changes in OC turnover along soil profiles
The close correlations between Δ14C values of NaF–NaOH extracts and residues along the soil profiles (see also Fig. S4) result in a parallel decline in Δ14C values of both fractions with soil depths. Not only was the same proportion of OC extracted across soil depths, but the absolute differences between Δ14C values of extracts and residues also remained constant. The observed constant extractability indicates that changes in Δ14C with depth are not driven by an increase in the stability of residual or extractable OC. This suggests that (1) the distribution of fast- and slower-cycling MOC is constant with depth and that (2) the shape of the 14C distribution of MOC (if we consider 14C in MOC to be a continuum) remains constant with depth. This is supported by the observation that residues of the H2O2 treatment, although much older on average, declined almost parallel to NaF–NaOH residues with soil depth. The factor that causes the decline in Δ14C values with soil depth is shifting the entire 14C distribution of MOC. This, along with the uniform extractability, suggests that subsoil and topsoil MOC are at comparable equilibriums with their environment, and the reasons for overall 14C declines with depth in soils are not due to changes in the character of mineral–organic matter stabilization mechanisms.
Previous studies showed that the oxidation-resistant proportion of bulk soil samples increased with soil depth (Butnor et al., 2017; Eusterhues et al., 2005). While the MOC analysed in this study showed only a small and insignificant trend in oxidation resistance with depth, we observed a stronger decline in Δ14C values of oxidation residues with depth relative to bulk MOC. Thus, oxidation resistance (i.e. chemical recalcitrance), rather than desorption resistance, could contribute to some extent to the formation of stable MOC with soil depth. The divergence of oxidized and residual Δ14C values as more OC is oxidized occurred across sites (Fig. S5), suggesting an overall trend of increasingly older OC with smaller amounts of OC remaining on the minerals.
5.1 Suitability of chemical fractionation schemes to study MOC stability
NaF–NaOH extraction and H2O2 oxidation results suggest that, rather than averaging over a wide distribution of ages, the majority of total MOC has very similar 14C contents, with only a small proportion (on average < 12 %) of total MOC being much older than the mean of the extracted or oxidized fractions. The different chemical fractionation schemes removed OC from the same pool, leaving increasingly old OC behind. Neither chemical oxidation nor the NaF–NaOH extractability of MOC were related to the Δ14C values of the removed OC. Therefore, the combined application of several fractionation schemes can help identify 14C distributions of samples (see Fig. S6), but their value for explaining site-specific differences in Δ14C values is limited.
5.2 Similarity of MOC extraction results across sites
The initial assumption of this study was that the stability of mineral-bound OC would be related to desorption and, thus, vary between soils with different mineral composition and under different land use conditions. We found that OC extracted in NaF–NaOH was indeed consistently younger than bulk MOC, suggesting that desorbed OC was more frequently exchanged than the older residue. The chemical uniformity of extracted MOC for three sites (two beech forests and one grassland) suggests a certain selection for specific OC molecules during MOC formation or that microbial processing of OC on mineral surfaces homogenizes MOC composition relative to original OM differences from litter and vegetation types. However, the Podzol site and the Chernozem site deviated from the pattern of the other results, indicating that factors including decomposition conditions affecting DOC or a specific fire history can affect the organic matter composition of MOC.
Despite differences in mineral and chemical composition, the extractability of MOC was uniform across non-cultivated soils and depths for the acidic to neutral central European soils studied. Based on these results, most extractable MOC is formed from specific interactive functional groups, including metal-coordinated hydroxyl groups on the mineral and carboxyl groups of OM. The minerals or OM molecules that these groups are attached to are interchangeable. As total MOC amounts were controlled by contents of pedogenic Al and Fe oxides across sites, extraction results may mostly reflect the response of oxides, which would overshadow potential mineral-specific differences in binding strengths observed in pure minerals in the laboratory. This supports the paradigm that oxides are more important than clay for OC storage in soil (Rasmussen et al., 2018) and could facilitate the modelling of MOC formation and turnover in the future.
5.3 Subsoil MOC stability
We observed no indication of the presence of stronger, less desorbable bonds between OC and minerals in subsoils than topsoils, and declines in the 14C content of extracts and extraction residues with depth paralleled each other. Consequently, subsoil OC was not better protected against desorption and, thus, potential subsequent degradation than topsoil OC. Although older, subsoil MOC can be equally vulnerable to changes in soil environmental conditions like temperature. Reduced NaF–NaOH extraction at the cropland site indicates that desorption may be explained in part by OC loading of minerals. A possible explanation for the missing depth gradient in MOC extraction is that subsoil MOC is typically at a similar equilibrium with the local conditions as the topsoil. This could be the case if OC input to undisturbed subsoils mostly occurs in hot spots, for example, via root inputs or along preferential flow paths, where OC binds to exposed mineral surfaces but bypasses surfaces located inside aggregates or away from such hot spots.
5.4 Alternative hypothesis for site- and depth-specific differences in 14C
Originally, we expected that the persistence of MOC would be related to different bond strengths expressed between OM and minerals and that these should vary with soil depth and mineralogy and could be identified by differing extractability into NaF–NaOH (Fig. 9). However, our results revealed an overall similarity in MOC composition and extractability across sites, despite site-specific variations in Δ14C values. As Δ14C values of bulk MOC and its fractions were therefore independent of OC resistance to desorption and soil mineralogy, site-specific differences are likely prompted by factors driving MOC accumulation and displacement, rather than by intrinsic differences in bond stabilities. Assuming that MOC is the product of DOC entering and leaving mineral surfaces, Δ14C values of MOC should be sensitive to site-specific differences in fluxes, composition, and Δ14C values of DOC (Fig. 9). These metrics should reflect differences in overall ecosystem properties and decomposition conditions, including litter chemistry, soil pH, microbial community composition, or climate, rather than soil mineralogy. Differences in depth profiles of Δ14C values would then be caused by variations in OC input by roots and DOC transport. Under conditions of lower direct litter input, vertical OC transport (e.g. the “cycling downwards” concept; Kaiser and Kalbitz, 2012) would shape the trends of Δ14C values with depth. Alternatively, differences in the stability of pedogenic oxide minerals themselves are also shaped by soil environmental factors.
For example, at the Podzol site Wetzstein, the thick organic layer (8–14 cm) overlying the topsoil could have contributed to old Δ14C values of total and extractable MOC in the uppermost mineral layer relative to the other undisturbed sites via the input of pre-aged DOC. Kindler et al. (2011) measured much higher DOC leaching rates from topsoils at Wetzstein than at Laqueuille or Hainich, despite only slightly higher subsoil leaching rates, suggesting that a large proportion of this mobilized topsoil DOC at Wetzstein was adsorbed in the subsoil. The absorption of leachate in the subsoil explains the shallower depth decline in 14C compared with the other two sites. Future research on the role of DOC formation, composition, and Δ14C values for accumulation and exchange rates of OC on mineral surfaces along soil profiles under field conditions will contribute to the understanding of the age distribution of MOC in soils.
Data will be published in the International Soil Radiocarbon Database (ISRAD).
All of the remaining material from the original MOC samples and the obtained fractions is stored at the Max Planck Institute for Biogeochemistry (MPI-BGC) and can be made available upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/bg-18-1241-2021-supplement.
MS, KK, AM, and ST designed the experiments, and AM carried them out. GH conducted the NMR analyses. MS prepared the paper with contributions from all co-authors.
The authors declare that they have no conflict of interest.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to the Routine Measurements and Analyses group (Roma; Ines Hilke and Birgit Fröhlich), the 14C Analytik group (Axel Steinhof and Heike Machts) of the Max-Planck Institute for Biogeochemistry, and Xiaomei Xu (UC Irvine) for their help with sample analyses.
The article processing charges for this open-access publication were covered by the Max Planck Society.
This paper was edited by Ji-Hyung Park and reviewed by two anonymous referees.
Aran, D., Gury, M., and Jeanroy, E.: Organo-metallic complexes in an Andosol: a comparative study with a Cambisol and Podzol, Geoderma, 99, 65–79, https://doi.org/10.1016/S0016-7061(00)00064-1, 2001.
Avneri-Katz, S., Young, R. B., McKenna, A. M., Chen, H., Corilo, Y. E., Polubesova, T., Borch, T., and Chefetz, B.: Adsorptive fractionation of dissolved organic matter (DOM) by mineral soil: Macroscale approach and molecular insight, Org. Geochem., 103, 113–124, https://doi.org/10.1016/j.orggeochem.2016.11.004, 2017.
Bruun, T. B., Elberling, B., and Christensen, B. T.: Lability of soil organic carbon in tropical soils with different clay minerals, Soil Biol. Biochem., 42, 888–895, https://doi.org/10.1016/j.soilbio.2010.01.009, 2010.
Bundt, M., Jaggi, M., Blaser, P., Siegwolf, R., and Hagedorn, F.: Carbon and nitrogen dynamics in preferential flow paths and matrix of a forest soil, Soil Sci. Soc. Am. J., 65, 1529–1538, https://doi.org/10.2136/sssaj2001.6551529x, 2001.
Butnor, J. R., Samuelson, L. J., Johnsen, K. H., Anderson, P. H., González Benecke, C. A., Boot, C. M., Cotrufo, M. F., Heckman, K. A., Jackson, J. A., Stokes, T. A., and Zarnoch, S. J.: Vertical distribution and persistence of soil organic carbon in fire-adapted longleaf pine forests, Forest Ecol. Manage., 390, 15–26, https://doi.org/10.1016/j.foreco.2017.01.014, 2017.
Chen, H., Koopal, L. K., Xiong, J., Avena, M., and Tan, W.: Mechanisms of soil humic acid adsorption onto montmorillonite and kaolinite, J. Colloid Interf. Sci., 504, 457–467, https://doi.org/10.1016/j.jcis.2017.05.078, 2017.
Chenu, C. and Stotzky, G.: Interactions between microorganisms and soil particles: an overview, in: Interactions between Soil Particles and Microorganisms: Impact on the Terrestrial Ecosystem, edited by: Huang, P. M., Bollag, J.-M., and Senesi, N., John Wiley & Sons, Ltd, Chichester, 3–40, 2002.
Chorover, J. and Amistadi, M. K.: Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces, Geochim. Cosmochim. Ac., 65, 95–109, https://doi.org/10.1016/S0016-7037(00)00511-1, 2001.
Cotrufo, M. F., Soong, J. L., Horton, A. J., Campbell, E. E., Haddix, M. L., Wall, D. H., and Parton, A. J.: Formation of soil organic matter via biochemical and physical pathways of litter mass loss, Nat. Geosci., 8, 776–779, https://doi.org/10.1038/ngeo2520, 2015.
Cotrufo, M. F., Ranalli, M. G., Haddix, M. L., Six, J., and Lugato, E.: Soil carbon storage informed by particulate and mineral-associated organic matter, Nat. Geosci., 12, 989–994, https://doi.org/10.1038/s41561-019-0484-6, 2019.
Eusterhues, K., Rumpel, C., Kleber, M., and Kogel-Knabner, I.: Stabilisation of soil organic matter by interactions with minerals as revealed by mineral dissolution and oxidative degradation, Org. Geochem., 34, 1591–1600, https://doi.org/10.1016/j.orggeochem.2003.08.007, 2003.
Eusterhues, K., Rumpel, C., and Kogel-Knabner, I.: Stabilization of soil organic matter isolated via oxidative degradation, Org. Geochem., 36, 1567–1575, https://doi.org/10.1016/j.orggeochem.2005.06.010, 2005.
Eusterhues, K., Neidhardt, J., Hadrich, A., Kusel, K., and Totsche, K. U.: Biodegradation of ferrihydrite-associated organic matter, Biogeochemistry, 119, 45–50, https://doi.org/10.1007/s10533-013-9943-0, 2014.
Gu, B., Schmitt, J., Chen, Z., Liang, L., and McCarthy, J. F.: Adsorption and desorption of natural organic matter on iron oxide: mechanisms and models, Environ. Sci. Technol., 28, 38–46, https://doi.org/10.1021/es00050a007, 1994.
Heckman, K., Lawrence, C. R., and Harden, J. W.: A sequential selective dissolution method to quantify storage and stability of organic carbon associated with Al and Fe hydroxide phases, Geoderma, 312, 24–35, https://doi.org/10.1016/j.geoderma.2017.09.043, 2018.
Helfrich, M., Flessa, H., Mikutta, R., Dreves, A., and Ludwig, B.: Comparison of chemical fractionation methods for isolating stable soil organic carbon pools, Eur. J. Soil Sci., 58, 1316–1329, https://doi.org/10.1111/j.1365-2389.2007.00926.x, 2007.
Hemingway, J. D., Rothman, D. H., Grant, K. E., Rosengard, S. Z., Eglinton, T. I., Derry, L. A., and Galy, V. V.: Mineral protection regulates long-term global preservation of natural organic carbon, Nature, 570, 228–231, https://doi.org/10.1038/s41586-019-1280-6, 2019.
Herold, N., Schoening, I., Michalzik, B., Trumbore, S., and Schrumpf, M.: Controls on soil carbon storage and turnover in German landscapes, Biogeochemistry, 119, 435–451, https://doi.org/10.1007/s10533-014-9978-x, 2014.
Jagadamma, S., Lal, R., Ussiri, D. A. N., Trumbore, S. E., and Mestelan, S.: Evaluation of structural chemistry and isotopic signatures of refractory soil organic carbon fraction isolated by wet oxidation methods, Biogeochemistry, 98, 29–44, https://doi.org/10.1007/s10533-009-9374-0, 2010.
Jones, D. L. and Edwards, A. C.: Influence of sorption on the biological utilization of two simple carbon substrates, Soil Biol. Biochem., 30, 1895–1902, https://doi.org/10.1016/s0038-0717(98)00060-1, 1998.
Kaiser, K. and Zech, W.: Release of natural organic matter sorbed to oxides and a subsoil, Soil Sci. Soc. Am. J., 63, 1157–1166, 1999.
Kaiser, K. and Guggenberger, G.: The role of DOM sorption to mineral surfaces in the preservation of organic matter in soils, Org. Geochem., 31, 711–725, https://doi.org/10.1016/S0146-6380(00)00046-2, 2000.
Kaiser, K. and Guggenberger, G.: Sorptive stabilization of organic matter by microporous goethite: sorption into small pores vs. surface complexation, Eur. J. Soil Sci., 58, 45–59, https://doi.org/10.1111/j.1365-2389.2006.00799.x, 2007.
Kaiser, K., Mikutta, R., and Guggenberger, G.: Increased stability of organic matter sorbed to ferrihydrite and goethite on aging, Soil Sci. Soc. Am. J., 71, 711–719, https://doi.org/10.2136/sssaj2006.0189, 2007.
Kaiser, K. and Kalbitz, K.: Cycling downwards - dissolved organic matter in soils, Soil Biol. Biochem., 52, 29–32, https://doi.org/10.1016/j.soilbio.2012.04.002, 2012.
Kalbitz, K., Schwesig, D., Rethemeyer, J., and Matzner, E.: Stabilization of dissolved organic matter by sorption to the mineral soil, Soil Biol. Biochem., 37, 1319–1331, https://doi.org/10.1016/j.soilbio.2004.11.028, 2005.
Kallenbach, C. M., Frey, S. D., and Grandy, A. S.: Direct evidence for microbial-derived soil organic matter formation and its ecophysiological controls, Nat. Commun., 7, 13630, https://doi.org/10.1038/ncomms13630, 2016.
Keil, R. G., Montluçon, D. B., Prahl, F. G., and Hedges, J. I.: Sorptive preservation of labile organic matter in marine sediments, Nature, 370, 549–552, https://doi.org/10.1038/370549a0, 1994.
Khomo, L., Trumbore, S., Bern, C. R., and Chadwick, O. A.: Timescales of carbon turnover in soils with mixed crystalline mineralogies, SOIL, 3, 17–30, https://doi.org/10.5194/soil-3-17-2017, 2017.
Kindler, R., Siemens, J., Kaiser, K., Walmsley, D. C., Bernhofer, C., Buchmann, N., Cellier, P., Eugster, W., Gleixner, G., Grunwald, T., Heim, A., Ibrom, A., Jones, S. K., Jones, M., Klumpp, K., Kutsch, W., Larsen, K. S., Lehuger, S., Loubet, B., McKenzie, R., Moors, E., Osborne, B., Pilegaard, K., Rebmann, C., Saunders, M., Schmidt, M. W. I., Schrumpf, M., Seyfferth, J., Skiba, U., Soussana, J. F., Sutton, M. A., Tefs, C., Vowinckel, B., Zeeman, M. J., and Kaupenjohann, M.: Dissolved carbon leaching from soil is a crucial component of the net ecosystem carbon balance, Glob. Change Biol., 17, 1167–1185, https://doi.org/10.1111/j.1365-2486.2010.02282.x, 2011.
Kleber, M., Mikutta, R., Torn, M. S., and Jahn, R.: Poorly crystalline mineral phases protect organic matter in acid subsoil horizons, Eur. J. Soil Sci., 56, 717–725, 10.1111/j.1365-2389.2005.00706.x, 2005.
Kleber, M., Eusterhues, K., Keiluweit, M., Mikutta, C., Mikutta, R., and Nico, P. S.: Chapter One – Mineral–Organic Associations: Formation, Properties, and Relevance in Soil Environments, in: Advances in Agronomy, edited by: Donald, L. S., Academic Press, 1–140, 2015.
Koarashi, J., Hockaday, W. C., Masiello, C. A., and Trumbore, S. E.: Dynamics of decadally cycling carbon in subsurface soils, J. Geophys. Res.-Biogeo., 117, 13, https://doi.org/10.1029/2012jg002034, 2012.
Kögel-Knabner, I., Guggenberger, G., Kleber, M., Kandeler, E., Kalbitz, K., Scheu, S., Eusterhues, K., and Leinweber, P.: Organo-mineral associations in temperate soils: Integrating biology, mineralogy, and organic matter chemistry, J. Plant Nutr. Soil Sci., 171, 61–82, https://doi.org/10.1002/jpln.200700048, 2008.
Kothawala, D. N., Roehm, C., Blodau, C., and Moore, T. R.: Selective adsorption of dissolved organic matter to mineral soils, Geoderma, 189, 334–342, https://doi.org/10.1016/j.geoderma.2012.07.001, 2012.
Lehmann, J. and Kleber, M.: The contentious nature of soil organic matter, Nature, 528, 60–68, https://doi.org/10.1038/nature16069, 2015.
Levin, I., Naegler, T., Kromer, B., Diehl, M., Francey, R. J., Gomez-Pelaez, A. J., Steele, L. P., Wagenbach, D., Weller, R., and Worthy, D. E.: Observations and modelling of the global distribution and long-term trend of atmospheric 14CO(2), Tellus B, 62, 26–46, 2010.
Liang, C., Schimel, J. P., and Jastrow, J. D.: The importance of anabolism in microbial control over soil carbon storage, Nat. Microbiol., 2, 17105, https://doi.org/10.1038/nmicrobiol.2017.105, 2017.
Mikutta, R., Kleber, M., Torn, M. S., and Jahn, R.: Stabilization of soil organic matter: Association with minerals or chemical recalcitrance?, Biogeochemistry, 77, 25–56, https://doi.org/10.1007/s10533-005-0712-6, 2006.
Mikutta, R., Mikutta, C., Kalbitz, K., Scheel, T., Kaiser, K., and Jahn, R.: Biodegradation of forest floor organic matter bound to minerals via different binding mechanisms, Geochim. Cosmochim. Ac., 71, 2569–2590, https://doi.org/10.1016/j.gca.2007.03.002, 2007.
Mikutta, R., Schaumann, G. E., Gildemeister, D., Bonneville, S., Kramer, M. G., Chorover, J., Chadwick, O. A., and Guggenberger, G.: Biogeochemistry of mineral-organic associations across a long-term mineralogical soil gradient (0.3–4100 kyr), Hawaiian Islands, Geochim. Cosmochim. Ac., 73, 2034–2060, https://doi.org/10.1016/j.gca.2008.12.028, 2009.
Mikutta, R., Kaiser, K., Dörr, N., Vollmer, A., Chadwick, O. A., Chorover, J., Kramer, M. G., and Guggenberger, G.: Mineralogical impact on organic nitrogen across a long-term soil chronosequence (0.3–4100 kyr), Geochim. Cosmochim. Ac., 74, 2142–2164, https://doi.org/10.1016/j.gca.2010.01.006, 2010.
Mikutta, R. and Kaiser, K.: Organic matter bound to mineral surfaces: Resistance to chemical and biological oxidation, Soil Biol. Biochem., 43, 1738–1741, https://doi.org/10.1016/j.soilbio.2011.04.012, 2011.
Möller, A., Kaiser, K., Amelung, W., Niamskul, C., Udomsri, S., Puthawong, M., Haumaier, L., and Zech, W.: Forms of organic C and P extracted from tropical soils as assessed by liquid-state 13C- and 31P-NMR spectroscopy, Soil Res., 38, 1017–1036, https://doi.org/10.1071/SR99111, 2000.
Oren, A. and Chefetz, B.: Successive sorption–desorption cycles of dissolved organic matter in mineral soil matrices, Geoderma, 189, 108–115, https://doi.org/10.1016/j.geoderma.2012.05.004, 2012.
Paul, E. A., Collins, H. P., and Leavitt, S. W.: Dynamics of resistant soil carbon of midwestern agricultural soils measured by naturally occurring 14C abundance, Geoderma, 104, 239–256, https://doi.org/10.1016/S0016-7061(01)00083-0, 2001.
Paul, S., Veldkamp, E., and Flessa, H.: Differential response of mineral-associated organic matter in tropical soils formed in volcanic ashes and marine Tertiary sediment to treatment with HCl, NaOCl, and Na4P2O7, Soil Biology and Biochemistry, 40, 1846–1855, https://doi.org/10.1016/j.soilbio.2008.03.008, 2008.
Plante, A. F., Pernes, M., and Chenu, C.: Changes in clay-associated organic matter quality in a C depletion sequence as measured by differential thermal analyses, Geoderma, 129, 186–199, https://doi.org/10.1016/j.geoderma.2004.12.043, 2005.
Poirier, N., Derenne, S., Balesdent, J., Mariotti, A., Massiot, D., and Largeau, C.: Isolation and analysis of the non-hydrolysable fraction of a forest soil and an arable soil (Lacadee, southwest France), Eur. J. Soil Sci., 54, 243–255, https://doi.org/10.1046/j.1365-2389.2003.00520.x, 2003.
Porras, R. C., Hicks Pries, C. E., McFarlane, K. J., Hanson, P. J., and Torn, M. S.: Association with pedogenic iron and aluminum: effects on soil organic carbon storage and stability in four temperate forest soils, Biogeochemistry, 133, 333–345, https://doi.org/10.1007/s10533-017-0337-6, 2017.
Porras, R. C., Hicks Pries, C. E., Torn, M. S., and Nico, P. S.: Synthetic iron (hydr)oxide-glucose associations in subsurface soil: Effects on decomposability of mineral associated carbon, Sci. Tot. Environ., 613, 342–351, https://doi.org/10.1016/j.scitotenv.2017.08.290, 2018.
Rasmussen, C., Southard, R. J., and Horwath, W. R.: Mineral control of organic carbon mineralization in a range of temperate conifer forest soils, Glob. Change Biol., 12, 834–847, https://doi.org/10.1111/j.1365-2486.2006.01132.x, 2006.
Rasmussen, C., Heckman, K., Wieder, W. R., Keiluweit, M., Lawrence, C. R., Berhe, A. A., Blankinship, J. C., Crow, S. E., Druhan, J. L., Hicks Pries, C. E., Marin-Spiotta, E., Plante, A. F., Schädel, C., Schimel, J. P., Sierra, C. A., Thompson, A., and Wagai, R.: Beyond clay: towards an improved set of variables for predicting soil organic matter content, Biogeochemistry, 137, 297–306, 10.1007/s10533-018-0424-3, 2018.
Rumpel, C. and Kögel-Knabner, I.: Deep soil organic matter-a key but poorly understood component of terrestrial C cycle, Plant Soil, 338, 143–158, https://doi.org/10.1007/s11104-010-0391-5, 2011.
Saidy, A. R., Smernik, R. J., Baldock, J. A., Kaiser, K., Sanderman, J., and Macdonald, L. M.: Effects of clay mineralogy and hydrous iron oxides on labile organic carbon stabilisation, Geoderma, 173, 104–110, https://doi.org/10.1016/j.geoderma.2011.12.030, 2012.
Schmidt, M. W. I., Torn, M. S., Abiven, S., Dittmar, T., Guggenberger, G., Janssens, I. A., Kleber, M., Kogel-Knabner, I., Lehmann, J., Manning, D. A. C., Nannipieri, P., Rasse, D. P., Weiner, S., and Trumbore, S. E.: Persistence of soil organic matter as an ecosystem property, Nature, 478, 49–56, https://doi.org/10.1038/nature10386, 2011.
Schrumpf, M., Kaiser, K., Guggenberger, G., Persson, T., Kögel-Knabner, I., and Schulze, E.-D.: Storage and stability of organic carbon in soils as related to depth, occlusion within aggregates, and attachment to minerals, Biogeosciences, 10, 1675–1691, https://doi.org/10.5194/bg-10-1675-2013, 2013.
Schrumpf, M. and Kaiser, K.: Large differences in estimates of soil organic carbon turnover in density fractions by using single and repeated radiocarbon inventories, Geoderma, 239, 168–178, https://doi.org/10.1016/j.geoderma.2014.09.025, 2015.
Singh, M., Sarkar, B., Biswas, B., Churchman, J., and Bolan, N. S.: Adsorption-desorption behavior of dissolved organic carbon by soil clay fractions of varying mineralogy, Geoderma, 280, 47–56, https://doi.org/10.1016/j.geoderma.2016.06.005, 2016.
Singh, M., Sarkar, B., Biswas, B., Bolan, N. S., and Churchman, G. J.: Relationship between soil clay mineralogy and carbon protection capacity as influenced by temperature and moisture, Soil Biol. Biochem., 109, 95–106, https://doi.org/10.1016/j.soilbio.2017.02.003, 2017.
Six, J., Callewaert, P., Lenders, S., De Gryze, S., Morris, S. J., Gregorich, E. G., Paul, E. A., and Paustian, K.: Measuring and understanding carbon storage in afforested soils by physical fractionation, Soil Sci. Soc. Am. J., 66, 1981–1987, https://doi.org/10.2136/sssaj2002.1981, 2002.
Steinhof, A., Altenburg, M., and Machts, H.: Sample Preparation at the Jena 14C Laboratory, Radiocarbon, 59, 815–830, https://doi.org/10.1017/RDC.2017.50, 2017.
Swanston, C. W., Torn, M. S., Hanson, P. J., Southon, J. R., Garten, C. T., Hanlon, E. M., and Ganio, L.: Initial characterization of processes of soil carbon stabilization using forest stand-level radiocarbon enrichment, Geoderma, 128, 52–62, https://doi.org/10.1016/j.geoderma.2004.12.015, 2005.
Torn, M. S., Trumbore, S. E., Chadwick, O. A., Vitousek, P. M., and Hendricks, D. M.: Mineral control of soil organic carbon storage and turnover, Nature, 389, 170–173, https://doi.org/10.1038/38260, 1997.
Trumbore, S. E., Vogel, J. S., and Southon, J. R.: AMS C-14 Measurements of fractionated soil organic-matter – an approach to deciphering the soil carbon-cycle, Radiocarbon, 31, 644–654, 1989.
Trumbore, S. E., Sierra, C. A., and Hicks Pries, C. E.: Radiocarbon Nomenclature, Theory, Models, and Interpretation: Measuring Age, Determining Cycling Rates, and Tracing Source Pools, in: Radiocarbon and Climate Change: Mechanisms, Applications and Laboratory Techniques, edited by: Schuur, E. A. G., Druffel, E., and Trumbore, S. E., Springer International Publishing, Cham, 45–82, 2016.
WRB, I. W. G.: World Reference Base for Soil Resources 2014, FAO, Rome, 2015.
Xu, X., Trumbore, S. E., Zheng, S., Southon, J. R., McDuffee, K. E., Luttgen, M., and Liu, J. C.: Modifying a sealed tube zinc reduction method for preparation of AMS graphite targets: Reducing background and attaining high precision, Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms, 259, 320–329, https://doi.org/10.1016/j.nimb.2007.01.175, 2007.
Young, A. W., Campbell, A. S., and Walker, T. W.: Allophane isolated from a podsol developed on a non-vitric parent material, Nature, 284, 46–48, https://doi.org/10.1038/284046a0, 1980.