the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Oct 2022
| 19 Oct 2022
Composition and niche-specific characteristics of microbial consortia colonizing Marsberg copper mine in the Rhenish Massif
Heiko Nacke
Elias Schliekmann
Andreas Reimer
Gernot Arp
Michael Hoppert
The Kilianstollen Marsberg (Rhenish Massif, Germany) has been extensively mined for copper ores, dating from early medieval period until 1945. The exposed organic-rich alum shale rocks influenced by the diverse mine drainages at an ambient temperature of 10 ∘C could naturally enrich biogeochemically distinct heavy metal resistant microbiota. This amplicon-sequence-based study evaluates the microbially colonized subterranean rocks of the abandoned copper mine Kilianstollen to characterize the colonization patterns and biogeochemical pathways of individual microbial groups. Under the selective pressure of the heavy metal contaminated environment at illuminated sites, Chloroflexi (Ktedonobacteria) and Cyanobacteria (Oxyphotobacteria) build up whitish–greenish biofilms. In contrast, Proteobacteria, Firmicutes and Actinobacteria dominate rocks around the uncontaminated spring water streams. The additional metagenomic analysis revealed that the heavy metal resistant microbiome was evidently involved in redox cycling of transition metals (Cu, Zn, Co, Ni, Mn, Fe, Cd, Hg). No deposition of metals or minerals, though, was observed by transmission electron microscopy in Ktedonobacteria biofilms which may be indicative for the presence of different detoxification pathways. The underlying heavy metal resistance mechanisms, as revealed by analysis of metagenome-assembled genomes, were mainly attributed to transition metal efflux pumps, redox enzymes, volatilization of Hg, methylated intermediates of As3+, and reactive oxygen species detoxification pathways.
- Article
(6326 KB) - Full-text XML
-
Supplement
(1481 KB) - BibTeX
- EndNote





The historic copper mining area Marsberg is situated on the northeastern edge of the Rhenish Schiefergebirge (Rhenish Massif) which is composed of Variscan folded rocks of Devonian and Carboniferous age (Urban et al., 1995). The Marsberg Upper Devonian sequence mainly consists of metamorphic clay shales, sandstones, siltstones, and carbonate rocks, whilst the Lower Carboniferous rocks contain a copper-rich black shale series (Siegmund et al., 2002). Investigations of the Marsberg copper ore deposits revealed insights in their geology, ore formation, and recent re-mineralizations (Stribrny, 1987). The copper-rich sediments formed about 380 million years ago in the Devonian on the southern edge of the Laurussia continent (America and Europe). The Marsberg copper deposit originated from tectonic movements which caused disintegration of the Lower Carboniferous alum shales and lydites and exposed the Upper Devonian rocks, resulting in fissures, faults, and breccia rich in metals (e.g. 7 %–16 % Cu), sulfides (0.5 %–3.8 %), carbonate carbon (0.35 %–2.43 %), and organic carbon (0.3 %–2.5 %) (Urban et al., 1995). In rock samples, mean contents of copper (81–1277 ppm) are found to be higher than those of other metals (Pb 36-417, Zn 78–660, Co 34–63, Ni 79–450, V 49–160, and Cr 45–193 ppm) (Urban et al., 1995). The Upper Devonian to Lower Carboniferous rocks are completely exposed in Marsberg Kilianstollen copper mine, offering conditions for formation of diverse secondary minerals and mine drainages. The most important sulfide ore minerals present in fault and fault-related breccia zones are described as chalcopyrite, neodigenite, chalcocite, bornite, and covellite (Stribrny, 1987). These minerals in the sediments all shape the prevailing biogeochemical conditions in the Marsberg mine waters. The biological and atmospheric oxidation of sulfides from pyrite (FeS2) or chalcopyrite (CuFeS2) could mobilize transition metals (Amin et al., 2018). The reduced transition metals (Cu, Fe, Mn) from fault-bound breccias and black alum shales are oxidized, resulting in copper-rich acidic mine waters with high concentrations of copper and iron, but also other dissolved ions (Fig. 1, green colour). In addition, karstic ground water enters the Kilianstollen (flowing in NE direction) that is enriched in calcium hydrogen carbonate from the Upper Permian Zechstein limestone, while sulfate is derived from gypsum and anhydrite of the same formation group (Fig. 1, blue colour). The calcium and sulfate levels in these mine waters can be higher than in fresh water, reaching up to of the values for sea water. These naturally flowing water streams of the Kilianstollen mine ranging from fresh water to heavy metal-enriched acidic leachate offer unique subsurface cold heavy metal-enriched habitats to study the colonized microbial communities under influence of various mine waters.
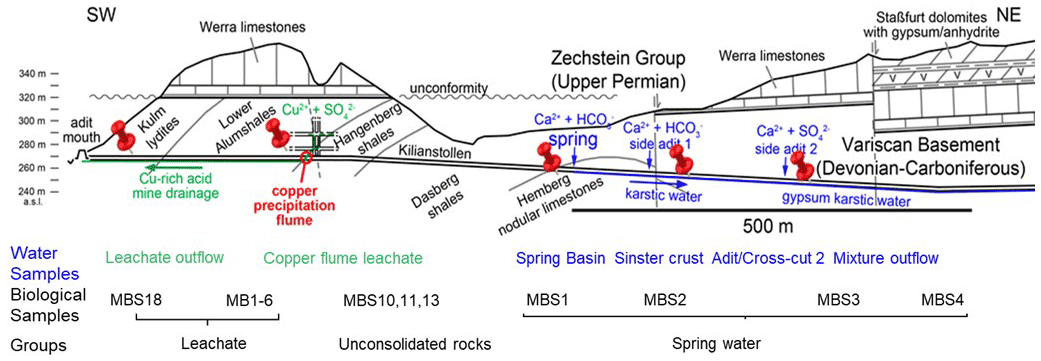
Figure 1Simplified geological section of the Kilianstollen (Marsberg) and formation of different ground waters. Folded Devonian–Carboniferous lydites, shales, and nodular limestones of the Variscan Basement are unconformably overlain by Upper Permian carbonates and evaporites of the Zechstein Group. The reduced transition metals (Cu, Fe, Mn) from fault-bound breccias and black shales are oxidized, resulting in copper-rich acidic mine waters (Emmerich, 1987), draining towards the adit entrance in SW direction. Some of the mine waters (flowing in NE direction) are enriched in calcium, hydrogen carbonate, and sulfate ions, depending on the rocks (limestone and gypsum) in contact. The geological section is based on (Îskar and William, 1936; Stribrny, 1987; Stribrny and Urban, 2000; Farrenschon et al., 2008) and an unpublished mining map. The red pins mark the location of the sampling sites along the mine drainage system for both water and biological samples collections. The biological samples were further classified into three groups based on their origin and nearby waters.
An ambient temperature of 10 ∘C, a relative humidity of 98 %, and an appropriate abundance of organics (2 %–10 %) in the alum shale, these organic-rich copper shales also provide microscale spaces for microbial colonization and aromatic compounds catabolism, perhaps because of their high content of kerogen, providing partially complex organic biodegradable compounds such as long-chain and polycyclic aromatic hydrocarbons, esters, organic acids, thiophenes, and metalloporphyrins (Dziewit et al., 2015). The availability of the soluble sulfate and transition metals ions from the nearby sulfuric waters sources (Silver and Walderhaug, 1992) are important in shaping an epilithic but also heavy metal and/or acid-tolerant bacterial community. Nevertheless, the emissions of the operational Marsberg mine railway diesel engine also provide another resource of organic compounds, and the regular visits of tourists, artificial ventilation, and illumination are some man-made impacts on the native microbial consortium.
In our previous study, enrichment cultures from slushy, iron- and manganese-rich secondary minerals from Kilianstollen, Marsberg were set up to observe the bioleaching from the copper slags. These were eventually enriched in iron and sulfur-oxidizing Acidithiobacillus ferrivorans, or Leptospirillum, indicating the presence of metal oxidizing communities (Amin et al., 2018). In this study, prokaryotic communities associated with the rocks around the Marsberg Kilianstollen mine waters were evaluated based on 16S rRNA gene amplicon sequencing to observe whether the mine waters enriched in transition metals may be toxic to microbial inhabitants or, conversely, support unique forms of metal respiration and enrich resistant microbial consortia under oligotrophic conditions. To elucidate further key processes involved in their resistance against high transition metal concentration and metabolism of the aromatic compounds, the metagenomically assembled genomes (MAGs) from a biofilm (MB1) abundant in Chloroflexi (Ktedonobacteria) nearby the copper-rich acidic mine waters were assembled and analysed for the genetic targets related to toxic Hg and As compounds reduction, Cu+ oxidation, heavy metal ions extrusion, dehalogenation, and hydrocarbon compounds catabolism. Understanding the selective pressure exerted by heavy metals on microbes and corresponding microbial resistance mechanisms could unveil their biogeochemical consequences and applications.
2.1 Sampling site
The rock samples colonized with soft biomass or biofilm around mine drainages of the Kilianstollen, Marsberg, Germany (51.453502∘ N, 8.861703∘ E) were collected under sterile precautions (Figs. 1, S1). Hammer and chisel were disinfected with 70 % ethanol prior to use and gloves were worn during sampling in order to reduce risks of contamination. The samples were taken from two locations in the alum shale region where mining activity was particularly high. For statistical analysis, the sampling sites shown in Fig. 2 [(MBS1–MBS4); (MB1–MB6, MBS18); (MBS10, 11, 13)] were divided into spring water, leachate, and unconsolidated rocks groups, respectively, based on the nearby drainage water bodies. A detailed description of the biological features and origin of the collected samples has been published previously (Arif et al., 2021b). Biofilm and water samples grouped as spring water samples (MBS1–MBS4) were taken along a small karst ground water stream that springs up in a cased basin and then naturally flows inside the Kilianstollen main gallery. Stream waters are typically enriched in calcium and hydrogen carbonate ions, and further downstream also in sulfate ions where water streams from crosscutting adits enter the main stream (Fig. 1). The biological samples under the influence of copper-rich acidic leachate waters, grouped as leachate samples (Figs. 1, 2), were taken from the mine outflow stream close to the entrance (MBS18) and from the immediate vicinity of a copper precipitation flume “Zement-Kupferplatte” (MB1–6 biofilms). The copper precipitation flume is an iron plate which is continuously flooded by a copper-rich acidic leachate seepage and the copper is being deposited electrochemically. The other samples from a wooden plank (MBS10) next to mine unconsolidated rocks (MBS11, 13) (Fig. 2) were grouped as unconsolidated samples. Samples collected for microscopic analysis were refrigerated until preparation. For extraction of genomic DNA, freshly collected samples were stored at −20 ∘C till further use. The samples (0.4 g each in triplicates) for genomic DNA extraction were obtained by scratching the biofilms of the rock piece and wooden plank with a sterile scalpel.

Figure 2Kilianstollen biological samples. Samples were taken from sites in the vicinity of a copper precipitation flume (a, b) that is drained actively with the leachate water, from a wooden plank (d) located just right to the copper flume, directly from the outflow stream (c) near the opening, and from rocky mine walls (e, f) exposed to the karstwater and pit water stream at the rear end crosscut of the Kilianstollen.
2.2 Hydro-chemical analysis
Mine drainage water around sampling sites was analysed on site using a WTW Multi 3430 device equipped with a WTW Tetracon 925 conductivity probe, a WTW FDO 925 probe for dissolved O2, and a WTW Sentix 940 electrode for temperature and pH (Xylem, Rye Brook, NY, USA). Calibration was performed with pH buffers 7.010 and 10.010 (HI6007, HI6010, Hanna Instruments, RI, USA). Redox potential was measured using a WTW 340i device equipped with a Schott PT61 redox electrode. To determine the anions and cations, water samples were collected without headspace in polyethylene bottles and for total alkalinity in Schott DURAN glass bottles (Schott, Mainz, Germany). Samples considered for cation analysis were filtered in separate 50 mL aliquots through 0.7 µm pore membrane filters and acidified with 100 µL HNO3 (65 %, Suprapure; Merck, Darmstadt, Germany). Total alkalinity (TA) was determined by acid-base titration within 24 h after sampling using a hand-held titration device and 1.6 N H2SO4 cartridges as titrant (Hach Lange GmbH, Düsseldorf, Germany). All other analysis were processed within 7 d after sampling.
Main cations (Ca, Mg, Na, and K) and anions (Cl, F, Br, SO4, and NO3) were analysed by ion chromatography with non-suppressed and suppressed conductivity detection (Metrohm 820 IC, Metrohm 883 Basic IC; Metrohm, Herisau), respectively. Minor element concentrations (Sr, Ba, Li, Rb, B) and trace metals were analysed either by inductively coupled plasma optical emission spectrometry (ICP–OES; Optima 3300 DV, Perkin–Elmer) or inductively coupled plasma mass spectrometry (ICP–MS; iCAP-Q, Thermo Fisher, Waltham, MA, USA) (see abundance graphs, https://doi.org/10.25625/DFFZ9R).
2.3 DNA extraction, 16S rRNA gene amplification, and amplicon sequencing
The microbial genomic DNA (gDNA) from 0.4 g scratched samples were extracted by using the DNeasy PowerSoil kit (Qiagen, Venlo, the Netherlands) as per manufacturer's instructions. In brief, the total gDNA released from cell lysis was treated for inhibitors removal and protein precipitation, then captured on and subsequently eluted from a silica membrane of a spin column. Blanks were also processed in addition to each sample to estimate DNA contamination. Following elution, the extracted gDNA was visually observed with 0.8 % agarose gel electrophoresis using TAE buffer, pH (8.3) (Sambrook and Russell, 2001), and quantified using a Nanodrop ND-1000 spectrophotometer (PeqLab, Erlangen, Germany). No gDNA contamination was measured in the blanks following the DNA extraction.
For Illumina MiSeq sequencing, V3–V4 hypervariable regions of 16S rRNA genes
were amplified via polymerase chain reaction (PCR) and tagged to 5′
overhang adapter sequences (underlined) with the aid of MiSeq 16S amplicon
PCR forward primer 341F 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCT
ACGGGNGGCWGCAG-3′ and reverse
primer 805R 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGA
CTACHVGGGTATCTAATCC-3′) (Klindworth et al., 2012). PCR
reaction mixture (Amin et al., 2018) was modified to obtain a final volume
of 50 µL in double-distilled nuclease-free water by mixing 5 × Phusion GC Buffer (10 µL), 10 µM forward and reverse primer (1.0 µL each), 10 mM dNTPs (1.0 µL), 5 % DMSO (, 2.5 µL), 50 mM MgCl2 (0.15 µL), 0.5 µL of 2 U µL−1 Phusion HF DNA
polymerase (Thermo-Fisher Scientific, Waltham, MA, USA) and 25 ng template
DNA (2.0 µL). The PCR profile comprised preheating at 94 ∘C
for 3 min followed by 25 cycles of heating at 94 ∘C for 45 s,
annealing at 60 ∘C for 45 s and extension at 72 ∘C for
30 s. The reaction ended with a final elongation step at 72 ∘C
for 5 min. After PCR amplification, the amplicons were visually assessed
with gel electrophoresis using 1.3 % () agarose in 1× TAE
buffer (Thermo-Fisher Scientific), pH 8.3 (Sambrook and Russell, 2001), and
photometrically quantified in a Nanodrop ND-1000 spectrophotometer (PeqLab).
The subsequent purification was performed with the GeneRead Size Selection
Kit (Qiagen) to remove primers and PCR reagents. After indexing of these PCR
amplicons using the Nextera XT DNA library prep kit (Illumina, San Diego,
CA, USA), paired-end sequencing was performed with an Illumina MiSeq
sequencer in collaboration with the Göttingen Genomics Laboratory.
2.4 Amplicon sequencing data processing
The Illumina amplicon sequencing data were processed online by employing the automated pipeline for analysis MetaAmp (http://ebg.ucalgary.ca/metaamp/, last access: 4 April 2020) (Dong et al., 2017). Using USEARCH software package, the demultiplexed Fastq format sequence files were assembled as paired-end reads (Edgar, 2010). The misaligned and mismatched reads and paired-end reads shorter than 350 bp length were discarded. Next, the primers were trimmed based on the Mothur software package (Schloss et al., 2009), and the reads without primers or with mismatched primer regions were removed. To minimize sequencing errors, the low-quality reads were scrapped using USEARCH. Dereplication, removal of singletons and chimeras, and clustering of pooled high-quality reads into operational taxonomic units (OTUs) on the basis of 97 % identity was done by UPARSE software (Edgar, 2013). The taxonomic status of the OTUs was assigned via Mothur by using the SILVA v138 as a reference (Glöckner, 2019). The taxonomic profile obtained from MetaAmp was further fed to the Microbiome online R-based server to plot respective graphs (Arndt et al., 2012). Based on the Bray–Curtis index, in principal coordinates analysis (PCoA), the data representing similarities of complex microbial communities were plotted into 2D and 3D graphs. Amplicon sequencing data have been published to the Sequence Read Archive (SRA) as SRR12876542-SRR12876555 under the project accession number PRJNA670497, as announced previously (Arif et al., 2021b).
16S rRNA gene sequences related to already published Ktedonobacteria and Actinobacteria strains were included for phylogenetic analysis. In a first step, sequences belonging to Ktedonobacteria or Actinobacteria-related OTUs were aligned with the already published sequences through MUSCLE, implemented in MEGA-X software, by using default settings (Stecher et al., 2020). Next, following the Kimura 2-parameter model, phylogenetic analyses and molecular evolutionary distances were calculated. The phylogenetic trees were constructed using the maximum likelihood algorithm and 1000 bootstrap samplings to test tree topology.
2.5 Microscopy
The morphological features of the sampled biofilms (MB1–6) from the leachate group were observed using a Motic SMZ-171 stereo microscope (Motic GmbH, Germany) equipped with a Canon A650 camera. For transmission electron microscopy (TEM), the biofilm MB2 specimens were washed with phosphate buffered saline (50 mM, pH 7.0–7.5), followed by fixation in 2 % () glutaraldehyde solution and incubation at 0 ∘C for 90 min. Subsequently, samples were dehydrated in a series of 15 %, 30 %, 50 %, 70 %, 95 %, and 100 % () aqueous ethanol solutions each for at least 30 min. After embedding samples with 66.6 % LR White resin (London Resin CO Ltd., UK) in ethanol at 25 ∘C for 2 h and overnight incubation in 100 % resin at 4 ∘C, the samples were polymerized for 12 h at 55 ∘C. The 80–100 nm ultrathin sections were cut with diamond knives (DDK, Wilmington, DE, USA) in Reichert Ultracut E ultramicrotome (Leica Biosystems, Wetzlar, Germany). The sections stabilized by formvar-coated 300 mesh copper grids (Plano GmbH, Wetzlar, Germany) were stained with Uranyl Acetate Replacement Stain (Electron Microscopy Sciences, Hatfield, PA, USA) for 20 min. Images were captured with a Gatan Orius 4 K camera attached to a Jeol 1011 electron microscope (Jeol GmbH, Munich, Germany) and processed with the 314 Gatan Digital Micrograph software (Gatan Inc., Pleasanton, USA) and Adobe CS2 Photoshop (Adobe Systems Inc., San José, Cal., USA).
2.6 Metabolic profiling based on metagenome-assembled genomes (MAGs)
Extracted DNA from one of the leachate biofilm samples, MB1, abundant in Ktedonobacteria (see Fig. 4) as a representative of leachate group, was submitted to the Göttingen Genomics Laboratory for shotgun metagenomic sequencing. The rationale behind selecting MB1 biofilm was to investigate the survival mechanisms that contributed to the high abundance of Ktedonobacteria around the toxic copper-rich leachate stream. The gDNA extraction followed by quality and quantity assessment was performed as described above. Illumina paired-end sequencing libraries were prepared using the Nextera DNA sample preparation kit and subsequently sequenced on a MiSeq system with the reagent kit v3 with 600 cycles (Illumina). For pre-processing of sequencing data, quality control, per-read quality pruning, read filtering, adapter trimming, and base correction, fastp v.0.19.4 (Chen et al., 2018) was used. The assembly of short read metagenomic data into metagenomic scaffolds was carried out by the metagenome assembler metaSPAdes v.3.14.0 (Nurk et al., 2017) with K-mers 21, 33, 55. Subsequently, bins were determined using MaxBin v.2.2.7 (Wu et al., 2015). CheckM v.1.1.2 was used to evaluate the MAGs quality by providing robust estimates of genome completeness and contamination (Parks et al., 2015). Each high-quality MAG was then annotated using PROKKA v1.14.5 (Seemann, 2014). Genome wide orthologous clusters across multiple species were determined with a web server, OrthoVenn v2 (Xu et al., 2019), which assigned the protein sequence data to a high-level summary of functional categories such as biological process, molecular function, and cellular component with GOSlim annotation and UniProt search. Finally, the Prokka output was analysed by using the PathoLogic (Karp et al., 2011) component of the Pathway Tools software v.23.5 (Karp et al., 2015) and the MetaCyc database v.23.5 (Caspi et al., 2019). The MAGs were classified taxonomically using GTDB-Tk v.1.0.2 and the Genome Taxonomy Database (GTDB) (release 89) (Chaumeil et al., 2020; Parks et al., 2019). The raw sequencing and assembly data have been already published in the SRA (SRR12886061) and Genbank (JADEYI000000000 and JADMIG000000000-JADMN000000000) under the project accession number PRJNA670497 (Arif et al., 2021a). The pathways maps of these MAGs showing their metabolic potentials and annotations can also be assessed at the Göttingen Research Online Database https://doi.org/10.25625/W9PWCX.
3.1 Physiochemical parameters of Kilianstollen mine waters
The physiochemical parameters (pH, electrical conductivity, redox potential) and ion concentrations were determined for the spring water samples (spring basin; source of karst water), and after the intermixing of an influent flow also rich in calcium and/or hydrogen carbonate (side adit 1), where a solid sinter crust was formed in the stream bed. Two other records of parameters were taken in a side stream rich in calcium and sulfate ions (side adit 2, “gypsum karstic water”), and after mixing with the karstic water of the mainstream. Further, two samples were taken from a copper flume leachate and a copper sulfate leachate outflow, draining towards the adit mouth (Fig. 1). The concentration of ions from transition metals Fe, Cu, Zn, and Mn, and Ca, Na, Cl, and SO were higher in these samples than in karst water samples (Fig. 3). The highest concentration of ions from Ni, Co, Fe, Cu, Zn, and Mn (0.62, 0.46, 35, 85, 2, and 4 mg L−1), was observed in the acidic copper flume leachate (pH 4.8) along with high concentrations of SO, Ca, Na, Cl, and NO (1950, 403, 415, 499, and 188 mg L−1) (Fig. 3a). Due to the metal precipitation, the concentrations of these transition metals towards the adit leachate outflow stream dropped to 0.2, 0.1, 2.9, 0.005, 0.8, and 1.3 mg L−1, respectively, and pH raised to 7.26. The heavy metal content in the spring water stream was considerably lower, i.e. in the range of 0.23 to 5.9 µg L−1. The efflux of the transition metal ions from the adit crosscut 2 drainage raised the heavy metal concentration of the stream water to the range of 3.8 to 262 µg L−1, particularly for Zn, Mn, Cu, and Ni (262, 76, 44, and 18 µg L−1). To understand the copper toxicity and homeostasis with respect to microbial consortia, biofilms growing at the rocky mine walls were investigated nearby these water bodies.
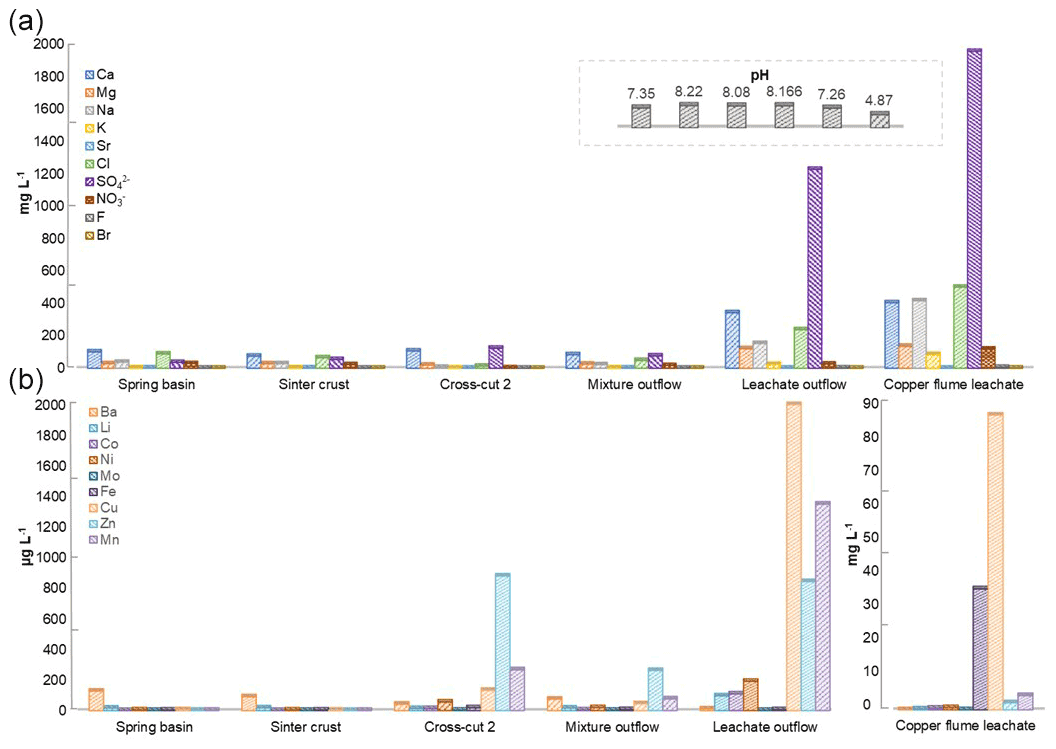
Figure 3Heavy metal content in Marsberg drainage mine waters. The concentration of major ions related to alkali and halogen groups (a) and heavy metals (b) have been compared. The insert shows the pH of each mine water in the same order as depicted for major ions. The copper flume heavy metal measurements were separately plotted as the copper and iron ion concentrations were exceptionally high. The spring basin stream continues to the sinter crust stream in the NE direction. The adit crosscut 2 mine drainage mixed with the sinter crust stream to form the mixture outflow stream. The copper flume and leachate outflow streams flow SW direction.
3.2 Alpha and beta diversity
The Shannon diversity indicated that unique OTUs (richness) were abundant in the MBS1–4 spring water stream samples, followed by the MBS10, 11, and 13 unconsolidated rocks, and MB1–6 and MBS18 leachate group samples (p=0.025 ANOVA) (Fig. 4). The alpha diversity index Chao1 index showed the same pattern when the sampling groups were statistically compared (p<0.0003 ANOVA, Table S1). Alpha diversity is high in the MBS1–4 samples, possibly due to moisture and neutral conditions from the adjacent spring water stream. With respect to the leachate samples group, low diversity indexes were observed as only a few adapted microbes could colonize, indicating a selective pressure due to extreme environmental conditions (Fig. 5a).
The principal coordinates analysis (PCoA) showed that the microbial communities of spring water, leachate, and unconsolidated rocks samples were distinct to each other (Fig. 4c, d). According to the Unifrac weighted algorithm (Lozupone et al., 2011) and analysis of molecular variance (AMOVA) nonparametric method (Table S2) (Mengoni and Bazzicalupo, 2002), the spring water and leachate samples were phylogenetically distinct to each other (p=0.034); conversely, the unconsolidated rocks microbiome was similar to both spring water and leachate groups (p>0.097). The unconsolidated rocks group may be the intermediate between the other groups in terms of diversity, and the diverse environmental conditions led to selection of different microorganisms. The hypothesis that the low pH and the high heavy metal concentration of mine water contributed to the Ktedonobacteria and Actinobacteria natural selection was further analysed with the canonical correspondence analysis (Fig. S2). The representative of leachate groups (MB1, MBS18) cluster closely with the low pH, high heavy metal concentration, and Ktedonobacteria abundance as compared to the spring water samples which showed that after mixing with the adit leachate, the major abundant taxa Proteobacteria (MBS1-2) shifted towards Actinobacteria (MBS3).

Figure 4Alpha and beta diversity. Shannon diversity index (a) and box plots (b) depict that the spring water samples have the highest alpha diversity. The PCoA 2D (c) and 3D (d) graphs indicated the unconsolidated rocks (+) microbial communities share similarity with both the spring water (*) and mine leachate (–) samples microbiome.
3.3 Distribution of bacterial taxa in Marsberg Kilian copper mine samples
The distribution of the predominant taxa at the collection sites varied drastically with the quality and type of nearby water sources (Fig. 5a). The most obvious sign of microbial growth, visible to the naked eye, were sub-aerial, whitish biofilms (MBS1–4) growing on the rocks, nearby the spring water stream. They were dominated by Proteobacteria (38 %) and Actinobacteria (21 %). The relative abundance of Proteobacteria declined, whereas the relative abundance of Actinobacteria increased gradually as the sampling site moves from spring water (MBS1) to karstic water containing heavy metal discharge influx (MBS3). In the leachate samples group, Chloroflexi (30 %), Cyanobacteria (23 %), and Actinobacteria (19 %) were abundant in the greenish–whitish biofilms (MB1–6) collected either in close vicinity of the copper flume leachate or directly from the heavy metal leachates streams (outflow water stream sample MBS18). Since these sites were more intensively illuminated with light bulbs than other sampling sites, this could have facilitated the growth of Cyanobacteria. The biofilms collected next to the copper flume from wooden plank (MBS10) and moist unconsolidated rocks (MBS11, 13) were enriched mainly in Actinobacteria (41 %) and Acidobacteria (20 %). When the three sample groups were compared in terms of abundant taxa, Chloroflexi and Cyanobacteria were significantly abundant in the leachate, while Firmicutes were relatively abundant in the spring water stream group samples and Actinobacteria and Acidobacteria seemed to be ubiquitous (p<0.05 DESeq2, Table S3).
At the class taxonomic rank, 5657 OTUs were identified across all samples comprising Actinobacteria (25 %), Ktedonobacteria (13 %), Oxyphotobacteria (12 %), Acidobacteria (9 %) Gammaproteobacteria (8 %), Deltaproteobacteria (5 %), Bacteroidia (5 %), and Alphaproteobacteria (3 %). MBS1–4 whitish biofilms growing nearby a spring water stream are dominated by Deltaproteobacteria and Gammaproteobacteria (48 %, MBS1), Bacilli (29 %, MBS2), and Actinobacteria (51 %, MBS3 and 25 %, MBS4) (Fig. 5a). Ktedonobacteria, being the most abundant class (26 %) in the leachate samples group (MB1–6 and MBS18), constituted 85 % of Chloroflexi. Oxyphotobacteria (a class within the phylum Cyanobacteria) and Actinobacteria also contributed 23 % and 16 % to the leachate group, respectively. Since the corresponding biofilms colonize rock surfaces in direct contact to the transition metal (Fe, Cu, Zn, and Mn) rich acidic mine drainage water (Figs. 1 and 2), it is hypothesized that the low pH and the high heavy metal concentration of mine water contributed to the Ktedonobacteria, Oxyphotobacteria, and Actinobacteria natural enrichment. Statistical analysis revealed that the classes Ktedonobacteria and Oxyphotobacteria were significantly abundant in the leachate group, while the Bacilli and Deltaproteobacteria in the spring water stream group and Actinobacteria and Acidobacteria classes in the unconsolidated rocks group were significantly abundant, as compared to other groups (p<0.05 DESeq2).

Figure 5Relative abundances of Marsberg Kilianstollen bacterial taxa. At phylum (a) and class (b) level, the taxonomy and relative abundance of the OTUs depicts the bacterial community composition and colonization at Kilianstollen sampling sites. The samples are shown in three groups based on their origin (unconsolidated rocks, leachate, and spring water). The classes showing less than 2 % relative abundance are not mentioned. Besides bacterial taxa, the selected primers also led to the detection of Crenarchaeota, Euryarchaeota, and Thaumarchaeota in low abundance. The excel data sheets are accessible at https://data.goettingen-research-online.de/dataset.xhtml?persistentId=doi:10.25625/DFFZ9R, last access: 4 May 2022.
3.4 Appearance of sampled biofilms and phylogenetic analysis
Eukaryotic microalgae and aerial mycelia could be observed in light microscopy images when the collected leachate samples group was visualized under the light microscope (Fig. 6). Interestingly, a nematode related to Poikilolaimus oxycercus was also found which colonizes the deep subsurface sites (Borgonie et al., 2019), and one unicellular alga (Coccomyxa subellipsoidea) was highly abundant, identified via 18S rRNA amplicon sequencing (Arif et al., 2021c); unpublished previous data). TEM micrographs also showed eukaryotic (algal) along with prokaryotic cells in the biofilm, indicating cohabitation. Mineral deposition around the microbial cell walls was not observed (Fig. 6), suggesting the inhabiting microbiota has employed some other pathways to cope with the heavy metal toxicity under low pH instead of metal precipitation. TEM micrographs of the MB3 biofilm from the leachate group also revealed the presence of sporulating hyphae which could be identified as either mycelia-like branched Ktedonobacteria, or Actinobacteria. Sporulation pattern (one spore per cell) and much higher relative abundance as compared to the Actinobacteria suggest the formation of the biofilm by Ktedonobacteria.
Marsberg Kilianstollen offers a large reservoir of uncultured strains, comprising numerous Chloroflexi representatives (Figs. S3, S4). The genus level-based analysis led to the identification of at least 10 distinct uncultured genera affiliated with the Ktedonobacteria class, and 80 % of them could be classified to the Ktedonobacteraceae family. Within Actinobacteria, 20 known genera were identified, whereby the Pseudonocardiaceae family represented 88 % actual abundance of these genera members. The most abundant genus, designated C0119, has been frequently assigned to Ktedonobacteria based on reference database comparisons (Dube et al., 2019; Paul Chowdhury et al., 2019). Nevertheless, the C0119 OTU have been shown to not cluster with Ktedonobacteria based upon phylogenetic analysis of Chloroflexi sequences (Glöckner, 2019; Jones et al., 2017) (Fig. S5). The unclassified C0119 strains might need to be classified into a new class of the cold climate adapted extremophiles belonging to the phylum Chloroflexi. In contrast, most of the Actinobacteria OTUs were classified as representatives of known genera such as Mycobacterium, Nocardioides, Micrococcus, Crossiella, Amycolatopsis, Nocardia, and Pseudonocardia which could be potentially interesting with respect to novel bioactive compounds, biodegradation, and biodeterioration pathways (Tiwari and Gupta, 2012; Kämpfer, 2010).

Figure 6Light and transmission electron microscopy of Marsberg Kilianstollen leachate group biofilms. The images (a–c) captured under the light microscope depict the aerial mycelium and green algal biomass along with the white bacterial biofilm. TEM micrographs (d) and (e) show the mycelial growth and spores formed at short branches along a hypha resembling Ktedonobacteria sporulation pattern (hypha and exospore are marked in d) and the large eukaryotic algal cells (e), respectively.
3.5 Key metabolic pathways to survive under extreme conditions
3.5.1 Metagenome-assembled genomes (MAGs)
Ktedonobacteria is a recently discovered hitherto poorly characterized class of Chloroflexi. The high abundance of Ktedonobacteria especially in a heavy metal contaminated environment (around copper leachate stream) implies specific survival mechanisms. To further elucidate these mechanisms, a biofilm sample MB1 as a representative of leachate group was selected for shotgun metagenomic sequencing based on the abundance of overall representative taxa of interest, such as Chloroflexi (Ktedonobacteria). Eight relatively complete MAGs (completeness, ≥ 89 %) were obtained, and subsequently proteins and metabolic pathways predicted. The genome sizes of the eight MAGs (designated Mberg 002, 006, 008, 009, 010, 011, 015, and 019), showing a contamination rate ≤ 10 %, ranged from 2.6 to 4.9 Mb. Furthermore, the number of identified genes ranged from 2516 to 4772. Based on phylogenetic analysis, the MAGs were assigned to Actinobacteria, Binatia, Deinococci, Chloroflexota, Dehalococcoidia, Chloroflexia, and Ktedonobacteria (Fig. S6).
3.5.2 Orthologous gene clusters
The preliminary comparison and analysis of GOSlim terms for core orthologous gene clusters for each MAG revealed unique survival pathways involved in extremophily (Fig. 7). The identified protein genes were grouped in 5540 distinct, 5395 orthologous, and unique 145 single-copy gene clusters. The shared orthologous gene ontology GO terms correspond to common cellular functions such as respiration and cell wall synthesis. These orthologous clusters contribute to various survival pathways such as aromatic and sulfur compounds metabolic processes, detoxification pathways for arsenic compounds and heavy metals ions transport, dehalogenation, and sporulation. The detected broad-spectrum heavy metal binding domains and associated proteins identified in the Prokka annotations are also important to survive high concentrations of heavy metals in the environment. The metal binding proteins are mostly enzymes which require transition metals Zn, Cu, Co, and Ni as cofactors to perform different biological processes. The cupredoxins superfamily proteins and domains were frequently found in shared clusters, and these contain type 1 Cu binding sites which are involved in oxidation reactions conferring resistance against Cu by various proteins (azurin, multicopper oxidases [MCO], laccases, and nitrosocyanin) (Argüllo et al., 2013; Vita et al., 2016; Donaire et al., 2002; Redinbo et al., 1994; Zaballa et al., 2012). Mberg010, assigned to Binatia, showed orthologous protein clusters related to aromatic compound degradation pathways. Mberg019, affiliated with Ktedonobacteria, has genes encoding unique plasma membrane proteins, identified in unshared clusters. All MAGs seem to mediate transition metal homeostasis, as metal binding proteins and transporters were frequently identified. The process of spore formation seems to be prevalent in all MAGs as GO terms for sporulation were observed in shared and unshared orthologous clusters. To study the MAGs with respect to heavy metal homeostasis, efflux systems, and detoxification and aromatic degradation pathways in more detail, pathway maps (https://doi.org/10.25625/W9PWCX) were generated based on Prokka outputs and subsequently inspected.
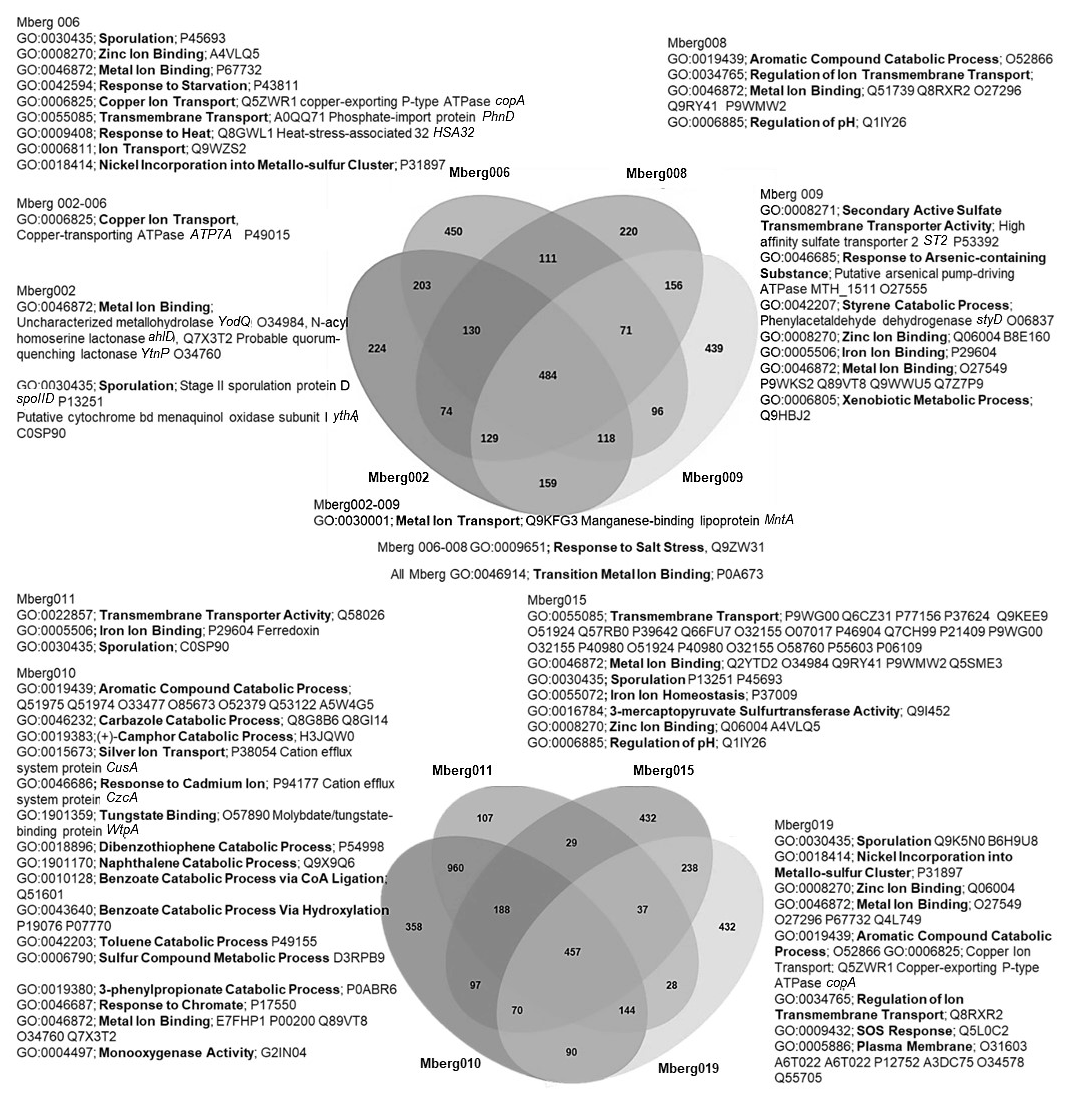
Figure 7Venn diagram displaying the distribution of shared orthologous clusters among the eight assembled genomes. The orthologous clusters of similar proteins have been allocated the GO terms based on same function or process. Only the GO terms related to metals, aromatic compound, sporulation, and pH regulations are mentioned to identify the microbial survival under extreme stress conditions. The closely matched proteins are identified as Swiss–Prot accession numbers, and names of some important hits are mentioned.
3.5.3 Heavy metal homeostasis and efflux systems
Transporters such as copper-exporting P-type and oxidation enzymes involved in transition metals homeostasis were identified in all selected MAGs (Fig. 8). The genes involved in copper homeostasis include copper-sensing transcriptional repressors CsoR and RicR, copper-exporting P-type ATPases CptA, ActP, and CopA, and oxidation enzymes, multicopper oxidases (MCOs), in all MAGs. Basically, upon Cu+ ion binding, the dissociation of CueR, CsoR, and RicR transcriptional repressors (Fu et al., 2014; Smaldone and Helmann, 2007) activates the copper regulon genes related to transporters CopA-like P-type ATPases, metallothioneins, or copper binding chaperons CopC, CopZ, and periplasmic multicopper oxidase MmcO and CueO (Osman et al., 2010; Shi et al., 2014). Under anaerobic conditions, the multicopper oxidase enzymes become inactive, and therefore another three-component channel or pore Cus complex controls Cu+ efflux through CusA, an inner membrane energy-driving channel which is attached to the outer membrane pore CusC through the periplasmic CusB protein (Outten et al., 2001).
The cutA locus, presumably involved in copper tolerance and homeostasis, was also characterized to affect tolerance levels to zinc, nickel, cobalt, and cadmium ions (Fong et al., 1995). The metalloregulatory transcriptional response to di- and multivalent heavy metal ions Cd2+, Pb2+, Bi3+, Zn2+, and Cu2+ is maintained by SmtB/ArsR family repressors CmtR and CadC (Busenlehner et al., 2003). MntH is a divalent metal cation transporter which displays broad substrate specificity and can regulate the intracellular accumulation of several divalent cations, including Mn2+, Cd2+, Co2+, Zn2+, and, to a lesser extent, Ni2+ and Cu2+ (Makui et al., 2000). CzcD, a heavy metal cation efflux transporter, mediates heavy metal resistance with respect to Cd2+, Co2+, and Zn2+ in the absence of the high resistance CzcCBA system (Nies, 2003; Anton et al., 1999; Papp-Wallace and Maguire, 2006).
Manganese homeostasis is maintained by a manganese efflux pump, MntP (Waters et al., 2011). A putative bacterial multicopper oxidase MoxA has been reported to compact the cellular Mn2+ toxicity through surface oxidation to the insoluble Mn3+ and Mn5+ oxides (Ridge et al., 2007; Zhang et al., 2015). HoxN, a high-affinity nickel transporter, facilitates the nickel translocation process as nickel permease (Wolfram et al., 1995). The influx of Mg2+, Ni2+ and Co2+ is coordinated via an ubiquitous divalent metal ion transporter CorA (Kersey et al., 2012), and efflux of these divalent metal ions is directed through CorC, a magnesium and cobalt efflux protein (Gibson et al., 1991). The homeostasis of the transition metals cobalt, nickel, and iron, and Ni2+ and Co2+ detoxification is also regulated by a nickel–cobalt exporter, designated RcnA, through efflux (Koch et al., 2007).
Cellular zinc uptake is regulated through the energy intensive import system ZnuABC, where ZnuA binds Zn2+ in the periplasmic space and docks Zn2+ to the membrane permease ZnuB. ZnuC finally catalyses ATP-dependent Zn2+ import into the cytosolic environment. In contrast, ZupT is involved in less energy intensive non-specific Zn2+ uptake along with the transition metals Fe2+, Co2+, Cd2+, and Mn2+ along the concentration gradient. Under high cytosolic divalent Zn2+, Cd2+, and Pb2+ concentrations, the MerR homologue, ZntR induces a Zn2+, Cd2+, and Pb2+ transporting P-type ATPase ZntA (Rensing and Mitra, 2007).
The oxyanions molybdate and tungstate are taken up through the membrane by the high-affinity ModABC molybdate system along with sulfate ions (Self et al., 2001; Xia et al., 2018; Maupin-Furlow et al., 1995). Sulfate and thiosulfate are taken up by sulfate permeases, carriers belonging to the SulT family, encoded by the cysPTWA operon, and SulP family members (inorganic anion uptake carriers) (Kertesz, 2001).
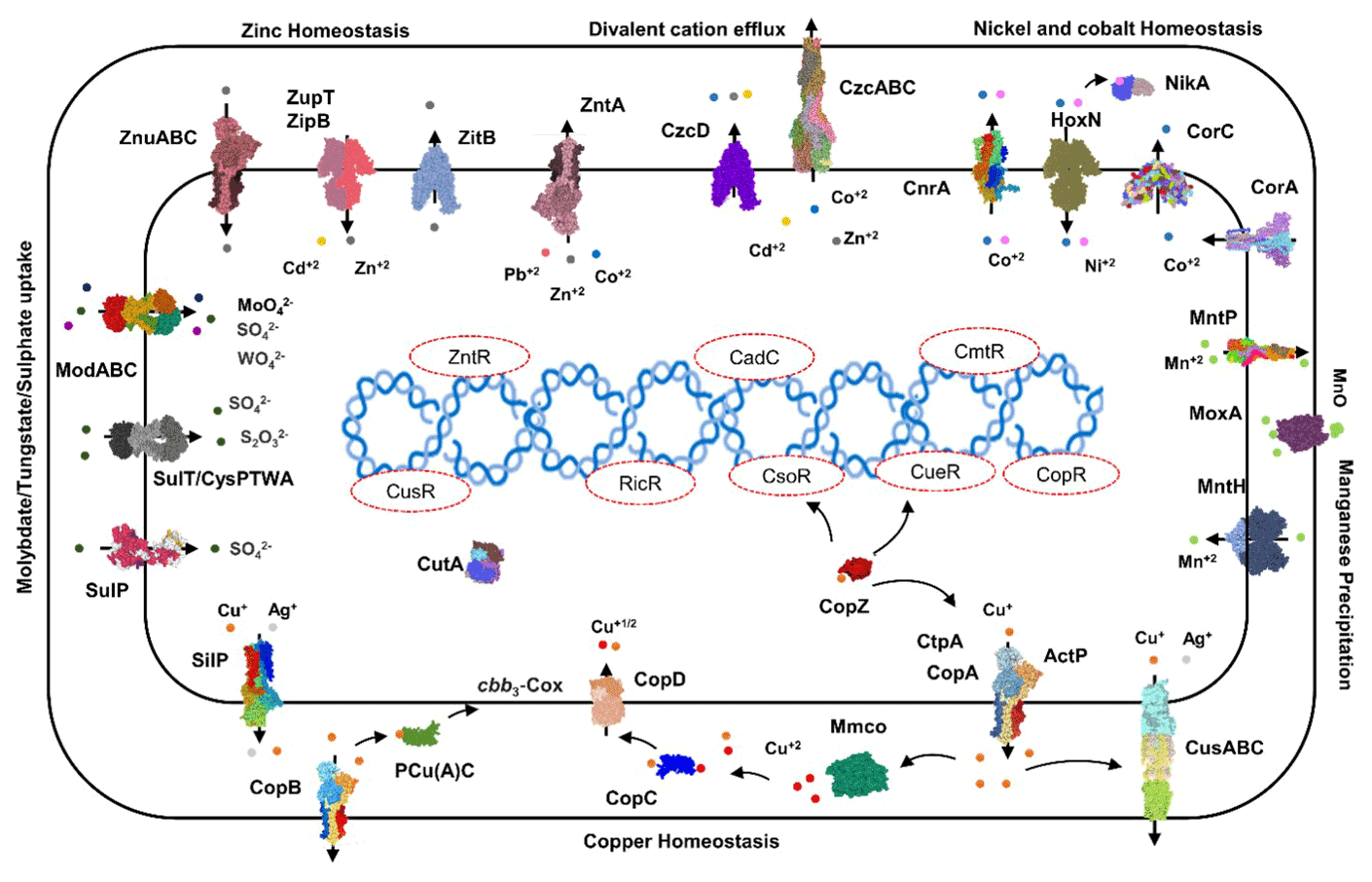
Figure 8Heavy metals transport channels and enzymes. The metalloregulatory transporters, enzymes, chaperones, and transcription factors form the specific protein–metal coordination complexes involved in the heavy metal transport, intracellular trafficking, storage, and detoxification are derived from the MAGs pathway maps (supplementary pdf files) and Prokka annotations (Arif et al., 2021a). Copper homeostasis is maintained through the efflux pumps CopA, CtpA, ActP, and CopB under aerobic conditions which remediate the high cellular Cu+ contents. The CopZ acts as allosteric switch to detect the excess copper ions, and activate the transcription of the CopABCD operon and multicopper oxidase MmcO through Cu-based inactivation of CsoR and CueR repressors. The periplasmic Cu+ is oxidized to the less soluble Cu2+ by MmcO and CopC that both bind Cu as storage and metallochaperone proteins, while PCu(A)C chaperones facilitate the biogenesis of the copper centre in the cytochrome oxidase.
3.5.4 Detoxification and aromatic degradation pathways
To successfully colonize the acid mine leachate downstream sites, microbes should harbour acid resistance, detoxification, and metabolizing systems as identified in all MAGs (Fig. 9). Arginine-dependent acid resistance relies on arginine decarboxylase SpeA, which decarboxylates arginine to produce agmatine (Tsai and Miller, 2013; Richard and Foster, 2004). Mercuric reductase MerB reduces divalent Hg2+ to volatile Hg (Silver and Hobman, 2007) and arsenate reductase ArsC reduces the arsenate (As5+) to arsenite (As3+) which is delivered to ATP-dependent anion pump ArsAB by the metallochaperone ArsD (Martin et al., 2001). The enzymes related to aromatic compounds (chlorinated phenols, benzoate, atrazine, cinnamate, biphenyl, phenylacetate, carbazole, catechol and 4-sulphocatechol, phenylethylamine, naphthalene, and 5-nitroanthranilate) degradation were mainly identified as hydroxylases, dioxygenases, dehydrogenases, epoxidase, etc. To detoxify reactive oxygen species (ROS), toxins, and antibiotics compounds with thiols, mycobacteria and some other Actinomycetales utilize mycothiol-mediated detoxification through Mca enzyme (Newton et al., 2000, 2008). Other detected important enzymes related to ROS detoxification were superoxide dismutase SOD and peroxidases (Broxton and Culotta, 2016).
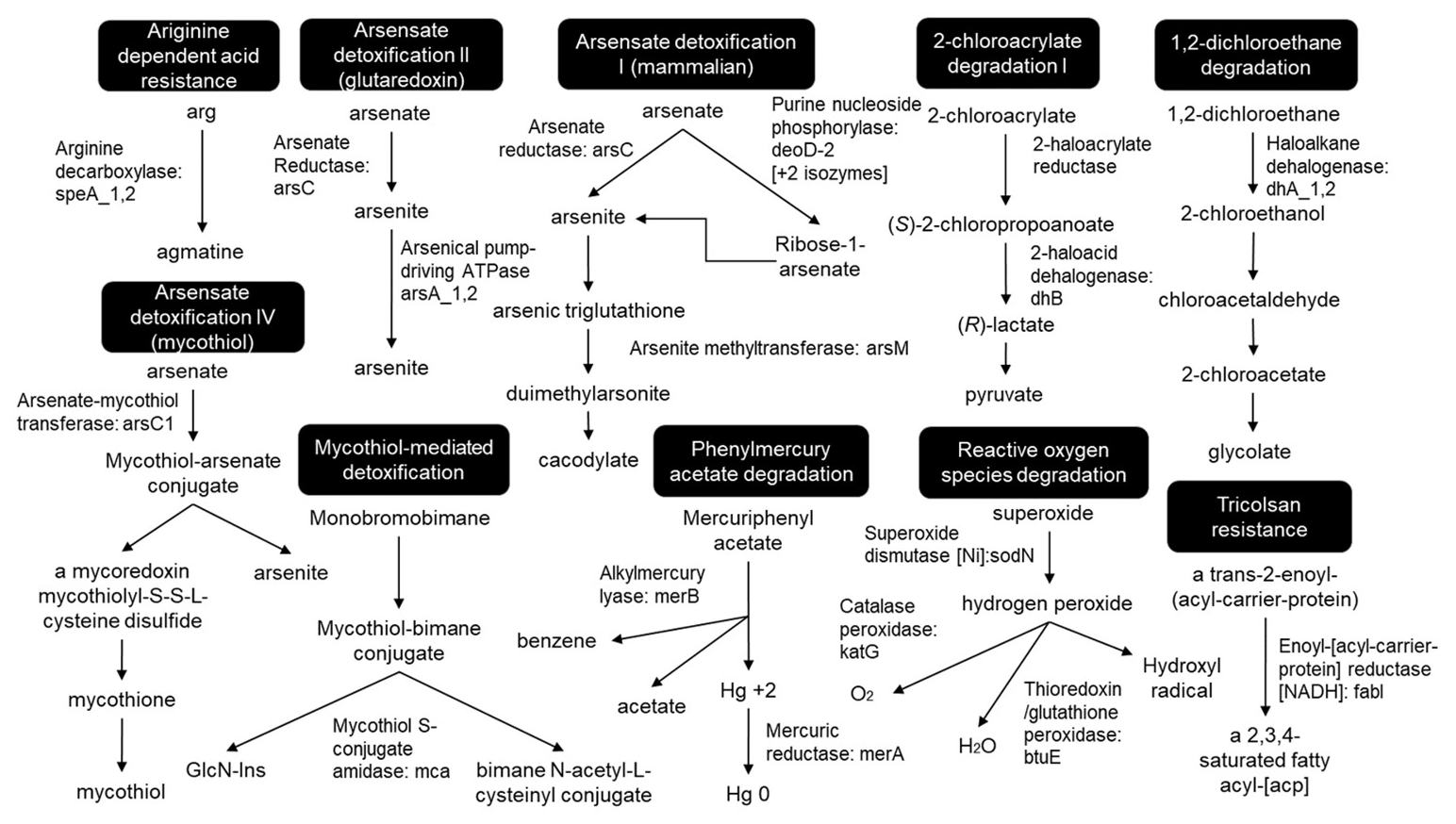
Figure 9Detoxification pathways. The pathways have been redrawn from the pathway maps of all MAGs, provided in the Supplement.
Generally, the acid mine drainage (AMD) habitats, which exceed most natural heavy metal concentrations, are dominated by prokaryotes with chemolithoautotrophic lifestyles, which produce sulfuric acid and are responsible for the mobilization of heavy metals from rocks through the oxidation of metal sulfide (Schippers and Sand, 1999). The present study aims to give a complete microbiome overview of Kilianstollen habitats influenced by mine drainage, where we could expect microbial metal reduction, deposition, and detoxification. To colonize the Marsberg mine subsurface habitat, the microbes must have developed resistance against heavy metals such as molybdenum, manganese, cobalt, zinc, and copper. Above optimal concentrations, copper confers its toxicity to microbes through its redox activity which catalyses a Fenton-like reaction, resulting in the generation of reactive oxygen species that may cause protein damage and lipid peroxidation (Macomber and Imlay, 2009) and destabilizing the iron–sulfur clusters via Fe(II) displacement in key enzymes prosthetic groups (Azzouzi et al., 2013; Chillappagari et al., 2010; Dupont et al., 2011). Thus, copper may inhibit the growth of common fast-growing Proteobacteria or Firmicutes such as Escherichia coli, Pseudomonas aeruginosa, Vibrio spp., Bacillus cereus, and Bacillus subtilis, even at micromolar concentrations (Gordon et al., 1994). This 16S rRNA gene V3–V4 amplicon sequencing based study of the Marsberg copper mine also observed the colonization of heavy metal tolerant microbes and inhibition of Proteobacteria and Firmicutes growth under the influence of copper toxicity. The colonization sites near the copper-rich leachate outflow stream such as a copper flume (MB1–6 biofilms) enrich copper resistant groups belonging to abundant phyla Chloroflexi (Ktedonobacteria, KD4-96), Actinobacteria, and Cyanobacteria (Oxyphotobacteria) as compared to the freshwater stream sites; these are mainly colonized by fastidious non-resistant Proteobacteria. KD4-96 has been frequently detected in soils contaminated with metals (aluminium, iron; Wegner and Liesack, 2017) and mining-affected waters (Kujala et al., 2018). Heavy metal resistance has been widely studied for Actinobacteria members especially in AMD environments (Schmidt et al., 2005). Ktedonobacteria become abundant in the thermophilic (Yabe et al., 2011, 2017b; Arif et al., 2021c), mesophilic (Cavaletti et al., 2006; Yabe et al., 2017a), and psychrophilic (Ghezzi et al., 2021; Barton et al., 2014) environments under heavy metal, temperature, nutrient, and/or hydrocarbon stress.
A metatranscriptomic study of an abandoned Pb–Zn mine (Coto Txomin, Spain) also determined that the heavy metal concentrations (up to 3220 and 97 g kg−1 of Pb, Cd, and Zn, respectively) and low pH (4–6) drastically influenced the soil microbial diversity, suppressed the relative abundance of Actinobacteria, Acidobacteria, and Alphaproteobacteria, and enhanced slow-growing metal and acid-tolerant taxa affiliated with Chloroflexi (Ktedonobacteria) (Epelde et al., 2015).
The same enrichment trend for Chloroflexi (Ktedonobacteria and KD4-96) was observed when a microcosm setup supplemented with acid mine drainage contaminated soil and cysteine hydrochloride was incubated for six months at 30 ∘C, which decreased the abundance of the major taxa Acidobacteria, Acidimicrobiia, Actinobacteria, and Thermoleophilia (Gupta et al., 2018). A high abundance of Ktedonobacteria at the downstream arsenic deposits of the acid sulfate hot spring Tengchong, China, suggests that these aerobic heterotrophic mesophiles and thermophiles may have been involved in arsenic reduction/toleration along with iron and sulfur oxidation cycles (Jiang et al., 2016). The Marsberg cold-adapted Ktedonobacteria also colonize the downstream acid mine leachate, indicating the ability to reduce or tolerate heavy metal ions. The co-occurrence of Cyanobacteria (Oxyphotobacteria) and Chloroflexi (Ktedonobacteria) could facilitate the growth of heterotrophs by providing carbon nutrients, and in return the heterotrophs may remediate the heavy metal contaminated sites, resulting in better microbial survival and colonization of the microbial consortium. The selective enrichment of Ktedonobacteria at Marsberg copper mine indicates the ability of these psychrophiles to inhabit cold environments and could be linked to heavy metal tolerance along with iron. The extreme conditions survival is attributed to the high metabolic plasticity of Ktedonobacteria, a diverse class ranging from thermophilic to mesophilic isolates, and the type stain Ktedonobacter racemifer has an unusually large genome of 13 Mbp, containing 9539 genes, 601 of which are transposases (Chang et al., 2011). The identified KO07665 based on KEGG analysis belonging to the Ktedonobacteriaceae codes for the copper resistance phosphate regulon response regulator CusR (Thomas Iv et al., 2020). The pathway map and annotation of the Marsberg Ktedonobacteria MAG019 also indicates a detection mechanism with respect to Cu+ by copper-sensing transcriptional repressor (RicR), oxidation to Cu2+ via multicopper oxidase (MCO), and finally export outside the cells through copper-exporting P-type ATPases (CopA, CtpA) and non-specific heavy metal cadmium, cobalt, and zinc H+-K+ antiporter (CzcD). When compared with the type strains of mesophilic K. racemifer and thermophilic T. hazakenisis, the cold-adapted MAG019 showed only 3.9 % unique orthologous shared clusters for heat shock, bacterial chemotaxis, protocatechuate branch of beta-ketoadipate pathway to degrade aromatic compounds, heavy metal resistance through Ton and Tol transport system, copper homeostasis, peptidoglycan biosynthesis, and DNA repair (Arif, 2021a).
High concentrations of heavy metal ions in the environment promote selection for heavy metal-resistant microbes, with either chromosomal or plasmid-level genes, maintaining heavy metal ion homeostasis inside cells (Rademacher and Masepohl, 2012). The organisms may reduce the sensitivity by employing the permeability barriers, by enhancement of active transport of metal ions from the cytoplasm (efflux) through a specific membrane transport system, by enzymatic detoxification, by reduction of metal ions by redox reactions, or by complexation of heavy metals resulting in extracellular and intracellular sequestration (Silver and Phung, 2005; Hobman et al., 2007). For all organisms in heavy metal environments, one or more of these adaptations are prerequisites for survival. This study investigated the pathways crucial for microbial survival nearby acid mine drainage, and all MAGs explicated had specialized adaptations to cope with heavy metal toxicity. Especially, copper homeostasis is maintained by copper-exporting P-type ATPases efflux pumps ActP, CptA, and CopA (Argüllo et al., 2013; Kim et al., 2008; Festa and Thiele, 2011), Cu+-specific metalloregulatory proteins (CsoR, RicR, Fu et al., 2014 and CopR, Villafane et al., 2011), oxidation of Cu+ to less toxic Cu2+ by the cupredoxins proteins superfamily (Rowland and Niederweis, 2013), and expression of metallochaperones and metallothioneins with Cu binding constants in the picomolar–femtomolar range (Rae et al., 1999; González-Guerrero and Argüello, 2008). The sensory cytosolic CopZ-like chaperones stimulate the transcription of several copper-stress related genes via trafficking the Cu+ to the DNA-bound CsoR and CueR and the inner membrane-localized channels, especially copper-exporting P-type ATP-ases such as ActP, CptA, and CopA for efflux to the extracytosolic periplasmic environment (Argüllo et al., 2013; Kim et al., 2008; Festa and Thiele, 2011; Novoa-Aponte et al., 2019). Mounting evidence also confirms the coexistence of the anaerobic Cus system with the aerobic CopA regulon in 44 % of Gammaproteobacteria group members (Hernández-Montes et al., 2012). The periplasmic CopC proteins maintain the bacterial copper homeostasis via binding both Cu+ and Cu2+ along with CopD inner membrane protein (Cha and Cooksey, 1993). PCuAc-like chaperone PccA required for the biogenesis of the copper centre assembly in the cbb3-type cytochrome c oxidases (CcO) have been demonstrated in various bacteria (Thompson et al., 2012; Andrei et al., 2020).
Since most of the heavy metal efflux pumps are ATPases, a constant supply of energy (as ATP) is met through converting various aromatic compounds to TCA cycle intermediates (Fuchs et al., 2011; Ghosal et al., 2016), in addition to several inorganic nutrients catabolism. To detoxify ROS, toxins, and antibiotic compounds, mycobacteria and some other Actinomycetales utilize mycothiol-mediated detoxification (Newton et al., 2000, 2008). Other important enzymes related to ROS detoxification are superoxide dismutase SOD and peroxidases which degrade the superoxide anion radicals (Broxton and Culotta, 2016). The arsRDABC operon codes for an ATP-dependent anion pump that confers resistance to arsenite, arsenate, antimonite, and tellurite (Rosen, 2002). Arsenite methyltransferase ArsM catalyses the formation of several volatile methylated intermediates from As3+, which eventually results in loss of arsenic from the cells through passive diffusion (Huang et al., 2018; Zeng et al., 2018). The mercuric reductase MerB reduces divalent Hg2+ to less bioavailable metallic mercury Hg vapour which is also volatilized under aerobic conditions and leaves the cells through passive diffusion (Silver and Hobman, 2007). These detoxification pathways also explain the lack of mineral or metals deposition as visible in TEM images as either oxidation of metallic ions to less soluble ionic form, or their reduction to volatile products makes them leave the bacteria through extrusion or diffusion under low pH. Hence, to inhabit the Marsberg acid mine leachate sites, the cold-adapted microbiome has the plasticity to express a wide range of heavy metal-specific enzymes which could either oxidize or reduce different metallic ions to neutralize their toxic effects, regulate their intracellular concentrations, and integrate the heavy metals as cellular components, cofactors for enzymatic functions, and protection against oxidative stress. Culturing of specific strains, especially Ktedonobacteria, may further clarify mechanisms of heavy metal resistance (exporter systems, metal chelators, bioorganic compounds enhancing metal precipitation). This might also give implications to mine waste treatment, bioremediation, and biomining.
The freshly collected samples from Marsberg copper mine (NE Rhenish Massif, Germany) were taken as a hitherto unexplored inventory of extremophilic organisms with largely unknown properties with respect to long-term survival, heavy metal tolerance, and degradation of complex organic compounds. The typical colonization patterns, mainly composed of Firmicutes, and Proteobacteria changed considerably towards uncultured Chloroflexi, including Ktedonobacteria representatives, when the sampling sites around the spring water stream shifted to the acid mine leachate outflow. The acid mine drainage with influx of heavy metals altered the composition, drastically reduced the richness and evenness of microbial communities, and exerted selective pressure towards resistance to metal contamination. Consequently, the microbiome has evolved various survival pathways related to aromatic and sulfur compounds metabolism, toxic arsenic compounds reduction, copper ions oxidation and heavy metal ions reduction and extrusion, dehalogenation, and sporulation.
The datasets related to relative abundance graphs and hydrochemistry are accessible at https://doi.org/10.25625/DFFZ9R (Arif, 2021b). The pathway maps of the MAGs can also be assessed at Göttingen Research Online, https://doi.org/10.25625/W9PWCX (Arif, 2021c).
The supplement related to this article is available online at: https://doi.org/10.5194/bg-19-4883-2022-supplement.
MH, NH and SA defined the methodology. SA, MH, ES, AR, GA and NH collected the water and rock samples. MH, SA and ES did the preliminary microscopic and amplicon sequencing analysis. SA and HN did the MAGs assembly and subsequent analysis. SA, ES, AR, GA and HN wrote down the original draft. The editing and further improvements were done by MH, SA, HN, AR and GA.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The support of Petra Ackermann, Gerhard Rosenkranz, and the “Marsberger Heimatbund e.V.” (Marsberg) is gratefully acknowledged. We would like to thank Deutscher Akademischer Austauschdienst (DAAD) for the provision of a doctoral research grant. The support by the Open Access Publication Funds of the University of Göttingen is also acknowledged.
This paper was edited by Jianming Xu and reviewed by Denise Akob and one anonymous referee.
Amin, N., Schneider, D., and Hoppert, M.: Bioleaching potential of bacterial communities in historic mine waste areas, Environ. Earth Sci., 77, 542, https://doi.org/10.1007/s12665-018-7714-x, 2018.
Andrei, A., Öztürk, Y., Khalfaoui-Hassani, B., Rauch, J., Marckmann, D., Trasnea, P.-I., Daldal, F., and Koch, H.-G.: Cu homeostasis in bacteria: the ins and outs, Membranes, 10, 242, https://doi.org/10.3390/membranes10090242, 2020.
Anton, A., Große, C., Reißmann, J., Pribyl, T., and Nies, D. H.: CzcD is a heavy metal ion transporter involved in regulation of heavy metal resistance in Ralstonia sp. strain CH34, J. Bacteriol., 181, 6876–6881, https://doi.org/10.1128/JB.181.22.6876-6881.1999, 1999.
Argüllo, J. M., Raimunda, D., and Padilla-Benavides, T.: Mechanisms of copper homeostasis in bacteria, Frontiers in cellular and infection microbiology, Front. Cell. Infect. Mi., 3, 73, https://doi.org/10.3389/fcimb.2013.00073, 2013.
Arif, S.: Organic-rich shales as archive and resource for microbial extremophiles, Georg-August-Universität Göttingen, http://hdl.handle.net/21.11130/00-1735-0000-0008-5906-E, last access: 10 October 2021a.
Arif, S.: Composition and Niche-Specific Characteristics of Microbial Consortia Colonizing Marsberg Copper Mine in the Rhenish Massif, GRO.data [data set], V1, https://doi.org/10.25625/DFFZ9R, 2021b.
Arif, S.: Pathway maps for Metagenome-assembled genome sequences from Marsberg Kilianstollen, GRO.data [data set], https://doi.org/10.25625/W9PWCX, 2021c.
Arif, S., Nacke, H., and Hoppert, M.: Metagenome-assembled genome sequences of a biofilm derived from Marsberg copper mine, Microbiol. Resour. Announ., 10, e01253-20, https://doi.org/10.1128/MRA.01253-20, 2021a.
Arif, S., Schliekmann, E., and Hoppert, M.: 16S rRNA Amplicon sequencing of microbial biofilms from Marsberg copper mine, Germany, Microbiol. Resour. Announ., 10, e01315-20, https://doi.org/10.1128/MRA.01315-20, 2021b.
Arif, S., Willenberg, C., Dreyer, A., Nacke, H., and Hoppert, M.: Sasso Pisano Geothermal Field environment harbours diverse Ktedonobacteria representatives and illustrates habitat-specific adaptations, Microorganisms, 9, 1402, https://doi.org/10.3390/microorganisms9071402, 2021c.
Arndt, D., Xia, J., Liu, Y., Zhou, Y., Guo, A. C., Cruz, J. A., Sinelnikov, I., Budwill, K., Nesbø, C. L., and Wishart, D. S.: METAGENassist: a comprehensive web server for comparative metagenomics, Nucl. Acid. Res., 40, 88–95, https://doi.org/10.1093/nar/gks497, 2012.
Azzouzi, A., Steunou, A. S., Durand, A., Khalfaoui-Hassani, B., Bourbon, M. l., Astier, C., Bollivar, D. W., and Ouchane, S.: Coproporphyrin III excretion identifies the anaerobic coproporphyrinogen III oxidase HemN as a copper target in the Cu+-ATPase mutant copA- of Rubrivivax gelatinosus, Mol. Microbiol., 88, 339–351, https://doi.org/10.1111/mmi.12188, 2013.
Barton, H. A., Giarrizzo, J. G., Suarez, P., Robertson, C. E., Broering, M. J., Banks, E. D., Vaishampayan, P. A., and Venkateswaran, K.: Microbial diversity in a Venezuelan orthoquartzite cave is dominated by the Chloroflexi (Class Ktedonobacterales) and Thaumarchaeota Group I, Front. Microbiol., 5, 615, https://doi.org/10.3389/fmicb.2014.00615, 2014.
Borgonie, G., Magnabosco, C., García-Moyano, A., Linage-Alvarez, B., Ojo, A. O., Freese, L. B., Van Jaarsveld, C., Van Rooyen, C., Kuloyo, O., Cason, E. D., Vermeulen, J., Pienaar, C., Van Heerden, E., Sherwood Lollar, B., Onstott, T. C., and Mundle, S. O. C.: New ecosystems in the deep subsurface follow the flow of water driven by geological activity, Sci. Rep., 9, 3310, https://doi.org/10.1038/s41598-019-39699-w, 2019.
Broxton, C. N. and Culotta, V. C.: SOD Enzymes and microbial pathogens: surviving the oxidative storm of infection, PLoS Pathog., 12, e1005295, https://doi.org/10.1371/journal.ppat.1005295, 2016.
Busenlehner, L. S., Pennella, M. A., and Giedroc, D. P.: The SmtB/ArsR family of metalloregulatory transcriptional repressors: structural insights into prokaryotic metal resistance, FEMS Microbiol. Rev., 27, 131–143, https://doi.org/10.1016/S0168-6445(03)00054-8, 2003.
Caspi, R., Billington, R., Keseler, I. M., Kothari, A., Krummenacker, M., Midford, P. E., Ong, W. K., Paley, S., Subhraveti, P., and Karp, P. D.: The MetaCyc database of metabolic pathways and enzymes – a 2019 update, Nucl. Acid. Res., 48, D445–D453, https://doi.org/10.1093/nar/gkz862, 2019.
Cavaletti, L., Monciardini, P., Bamonte, R., Schumann, P., Rohde, M., Sosio, M., and Donadio, S.: New lineage of filamentous, spore-forming, gram-positive bacteria from soil, Appl. Environ. Microbiol., 72, 4360–4369, https://doi.org/10.1128/AEM.00132-06, 2006.
Cha, J.-S. and Cooksey, D. A.: Copper hypersensitivity and uptake in Pseudomonas syringae containing cloned components of the copper resistance operon, Appl. Environ. Microbiol., 59, 1671–1674, https://doi.org/10.1128/aem.59.5.1671-1674.1993, 1993.
Chang, Y.-J., Land, M., Hauser, L., Chertkov, O., Del Rio, T. G., Nolan, M., Copeland, A., Tice, H., Cheng, J.-F., and Lucas, S.: Non-contiguous finished genome sequence and contextual data of the filamentous soil bacterium Ktedonobacter racemifer type strain (SOSP1-21 T), Stand. Genomic Sci., 5, 97–111, https://doi.org/10.4056/sigs.2114901, 2011.
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H.: GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database, Bioinformatics, 36, 1925–1927, https://doi.org/10.1093/bioinformatics/btz848, 2020.
Chen, S., Zhou, Y., Chen, Y., and Gu, J.: fastp: an ultra-fast all-in-one FASTQ preprocessor, Bioinformatics, 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560, 2018.
Chillappagari, S., Seubert, A., Trip, H., Kuipers, O. P., Marahiel, M. A., and Miethke, M.: Copper stress affects iron homeostasis by destabilizing iron-sulfur cluster formation in Bacillus subtilis, J. Bacteriol., 192, 2512–2524, https://doi.org/10.1128/JB.00058-10, 2010.
Donaire, A., Jiménez, B., Fernández, C. O., Pierattelli, R., Niizeki, T., Moratal, J.-M., Hall, J. F., Kohzuma, T., Hasnain, S. S., and Vila, A. J.: Metal–ligand interplay in blue copper proteins studied by 1H NMR Spectroscopy: Cu (II)-pseudoazurin and Cu (II)-rusticyanin, J. Am. Chem. Soc., 124, 13698–13708, https://doi.org/10.1021/ja0267019, 2002.
Dong, X., Kleiner, M., Sharp, C. E., Thorson, E., Li, C., Liu, D., and Strous, M.: Fast and simple analysis of MiSeq amplicon sequencing data with MetaAmp, Front. Microbiol., 8, 1461, https://doi.org/10.1101/131631, 2017.
Dube, J. P., Valverde, A., Steyn, J. M., Cowan, D. A., and Van der Waals, J. E.: Differences in bacterial diversity, composition and function due to long-term agriculture in soils in the eastern free State of South Africa, Diversity, 11, 61, https://doi.org/10.3390/d11040061, 2019.
Dupont, C. L., Grass, G., and Rensing, C.: Copper toxicity and the origin of bacterial resistance – new insights and applications, Metallomics, 3, 1109–1118, 2011.
Dziewit, L., Pyzik, A., Szuplewska, M., Matlakowska, R., Mielnicki, S., Wibberg, D., Schlüter, A., Pühler, A., and Bartosik, D.: Diversity and role of plasmids in adaptation of bacteria inhabiting the Lubin copper mine in Poland, an environment rich in heavy metals, Front. Microbiol., 6, 152, https://doi.org/10.1039/c1mt00107h, 2015.
Edgar, R. C.: Search and clustering orders of magnitude faster than BLAST, Bioinformatics, 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461, 2010.
Edgar, R. C.: UPARSE: highly accurate OTU sequences from microbial amplicon reads, Nat. Methods, 10, 996–998, https://doi.org/10.1038/nmeth.2604, 2013.
Emmerich, H. P. H., A: Sekundärmineralbildung aus Grubenwässern im Kupferbergwerk Niedermarsberg [Secondary mineral formation from mine water in the Niedermarsberg copper mine], Der Aufschluss, 38, 149–156, 1987.
Epelde, L., Lanzen, A., Blanco, F., Urich, T., and Garbisu, C.: Adaptation of soil microbial community structure and function to chronic metal contamination at an abandoned Pb-Zn mine, FEMS Microbiol. Ecol., 91, 1–11, https://doi.org/10.1093/femsec/fiu007, 2015.
Farrenschon, J., Oesterreich, B., Blumenstein, S., Holzinger, M., and Wrede, V.: Geologische Karte von Nordrhein-Westfalen 1:25 000, Erläuterungen zu Blatt 4519 Marsberg, Geological Map of North Rhine-Westphalia, Explanatory notes to sheet 4519 Marsberg, https://www.gd.nrw.de/pr_kd_geologische-karte-25000.php (last access: 2 February 2021), 2008.
Festa, R. A. and Thiele, D. J.: Copper: an essential metal in biology, Curr. Biol., 21, R877–R883, https://doi.org/10.1016/j.cub.2011.09.040, 2011.
Fong, S. T., Camakaris, J., and Lee, B. T. O.: Molecular genetics of a chromosomal locus involved in copper tolerance in Escherichia coli K-12, Mol. Microbiol., 15, 1127–1137, https://doi.org/10.1111/j.1365-2958.1995.tb02286.x, 1995.
Fu, Y., Chang, F.-M. J., and Giedroc, D. P.: Copper transport and trafficking at the host-bacterial pathogen interface, Accounts Chem. Res., 47, 3605–3613, https://doi.org/10.1021/ar500300n, 2014.
Fuchs, G., Boll, M., and Heider, J.: Microbial degradation of aromatic compounds – from one strategy to four, Nat. Rev. Microbiol., 9, 803–816, https://doi.org/10.1038/nrmicro2652, 2011.
Ghezzi, D., Sauro, F., Columbu, A., Carbone, C., Hong, P. Y., Vergara, F., De Waele, J., and Cappelletti, M.: Transition from unclassified Ktedonobacterales to Actinobacteria during amorphous silica precipitation in a quartzite cave environment, Sci. Rep., 11, 1–18, https://doi.org/10.1038/s41598-021-83416-5, 2021.
Ghosal, D., Ghosh, S., Dutta, T. K., and Ahn, Y.: Current State of Knowledge in Microbial Degradation of Polycyclic Aromatic Hydrocarbons (PAHs): A Review, Front. Microbiol., 7, 1369, https://doi.org/10.3389/fmicb.2016.01369, 2016.
Gibson, M. M., Bagga, D. A., Miller, C. G., and Maguire, M. E.: Magnesium transport in Salmonella typhimurium: the influence of new mutations conferring CO resistance on the CorA Mg transport system, Mol. Microbiol., 5, 2753–2762, https://doi.org/10.1111/j.1365-2958.1991.tb01984.x, 1991.
Glöckner, F. O.: The SILVA Database Project: An ELIXIR core data resource for high-quality ribosomal RNA sequences, Biodiv. Inf. Sci. Stand., 3, e36125, https://doi.org/10.3897/biss.3.36125, 2019.
González-Guerrero, M. and Argüello, J. M.: Mechanism of Cu+-transporting ATPases: soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites, P. Natl. Acad. Sci. USA, 105, 5992–5997, https://doi.org/10.1073/pnas.0711446105, 2008.
Gordon, A. S., Howell, L. D., and Harwood, V.: Responses of diverse heterotrophic bacteria to elevated copper concentrations, Can. J. Microbiol., 40, 408–411, https://doi.org/10.1139/m94-067, 1994.
Gupta, A., Dutta, A., Sarkar, J., Panigrahi, M. K., and Sar, P.: Low-abundance members of the Firmicutes facilitate bioremediation of soil impacted by highly acidic mine drainage from the Malanjkhand copper project, India, Front. Microbiol., 9, 2882, https://doi.org/10.3389/fmicb.2018.02882, 2018.
Hernández-Montes, G., Argüello, J. M., and Valderrama, B.: Evolution and diversity of periplasmic proteins involved in copper homeostasis in gamma proteobacteria, BMC Microbiol., 12, 249, https://doi.org/10.1186/1471-2180-12-249, 2012.
Hobman, J. L., Yamamoto, K., and Oshima, T.: Transcriptomic responses of bacterial cells to sublethal metal ion stress, in: Molecular microbiology of heavy metals, Springer, 73–115, https://doi.org/10.1007/7171_2006_074, 2007.
Huang, K., Xu, Y., Packianathan, C., Gao, F., Chen, C., Zhang, J., Shen, Q., Rosen, B. P., and Zhao, F.-J.: Arsenic methylation by a novel ArsM As(III) S-adenosylmethionine methyltransferase that requires only two conserved cysteine residues, Mol. Microbiol., 107, 265–276, https://doi.org/10.1111/mmi.13882, 2018.
Îskar, G. and William, P.: Erläuterungen zur geologischen Karte von Preußen und benachbarten deutschen Ländern, Blatt 4519 Marberg, Geological map of Prussia and neighboring German countries, 4519 Marsberg, https://katalog.slub-dresden.de/id/0-1390418065 (last access: 2 February 2021), 1936.
Jiang, Z., Li, P., Jiang, D., Dai, X., Zhang, R., Wang, Y., and Wang, Y.: Microbial community structure and arsenic biogeochemistry in an acid vapor-formed spring in Tengchong Geothermal Area, China, PloS One, 11, e0146331, https://doi.org/10.1007/7171_2006_074, 2016.
Jones, D. S., Lapakko, K. A., Wenz, Z. J., Olson, M. C., Roepke, E. W., Sadowsky, M. J., Novak, P. J., and Bailey, J. V.: Novel microbial assemblages dominate weathered sulfide-bearing rock from copper-nickel deposits in the Duluth complex, Minnesota, USA, Appl. Environ. Microbiol., 83, e00909-17, https://doi.org/10.1128/AEM.00909-17, 2017.
Karp, P. D., Latendresse, M., and Caspi, R.: The pathway tools pathway prediction algorithm, Stand. Genomic Sci., 5, 424–429, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3368424/ (last access: 27 September 2022), 2011.
Karp, P. D., Latendresse, M., Paley, S. M., Krummenacker, M., Ong, Q. D., Billington, R., Kothari, A., Weaver, D., Lee, T., Subhraveti, P., Spaulding, A., Fulcher, C., Keseler, I. M., and Caspi, R.: Pathway Tools version 19.0 update: software for pathway/genome informatics and systems biology, Brief. Bioinform., 17, 877–890, https://doi.org/10.1093/bib/bbv079, 2015.
Kersey, C. M., Agyemang, P. A., and Dumenyo, C. K.: CorA, the magnesium/nickel/cobalt transporter, affects virulence and extracellular enzyme production in the soft rot pathogen Pectobacterium carotovorum, Mol. Plant Pathol., 13, 58–71, https://doi.org/10.1111/j.1364-3703.2011.00726.x, 2012.
Kertesz, M. A.: Bacterial transporters for sulfate and organosulfur compounds, Res. Microbiol., 152, 279–290, https://doi.org/10.1016/s0923-2508(01)01199-8, 2001.
Kim, B.-E., Nevitt, T., and Thiele, D. J.: Mechanisms for copper acquisition, distribution and regulation, Nat. Chem. Biol., 4, 176–185, https://doi.org/10.1038/nchembio.72, 2008.
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., and Glöckner, F. O.: Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies, Nucl. Acid. Res., 41, e1, https://doi.org/10.1093/nar/gks808, 2012.
Koch, D., Nies, D. H., and Grass, G.: The RcnRA (YohLM) system of Escherichia coli: a connection between nickel, cobalt and iron homeostasis, Biometals, 20, 759–771, https://doi.org/10.1007/s10534-006-9039-6, 2007.
Kämpfer, P.: Actinobacteria, in: Handbook of Hydrocarbon and Lipid Microbiology, edited by: Timmis, K. N., Springer Berlin Heidelberg, Berlin, Heidelberg, 1819–1838, https://doi.org/10.1007/978-3-540-77587-4_133, 2010.
Kujala, K., Mikkonen, A., Saravesi, K., Ronkanen, A.-K., and Tiirola, M.: Microbial diversity along a gradient in peatlands treating mining-affected waters, FEMS Microbiol. Ecol., 94, fiy145, https://doi.org/10.1093/femsec/fiy145, 2018.
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., and Knight, R.: UniFrac: an effective distance metric for microbial community comparison, ISME J., 5, 169–172, https://doi.org/10.1038/ismej.2010.133, 2011.
Macomber, L. and Imlay, J. A.: The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity, P. Natl. Acad. Sci. USA, 106, 8344–8349, https://doi.org/10.1073/pnas.0812808106, 2009.
Makui, H., Roig, E., Cole, S. T., Helmann, J. D., Gros, P., and Cellier, M. F. M.: Identification of the Escherichia coli K-12 Nramp orthologue (MntH) as a selective divalent metal ion transporter, Mol. Microbiol., 35, 1065–1078, https://doi.org/10.1046/j.1365-2958.2000.01774.x, 2000.
Martin, P., DeMel, S., Shi, J., Gladysheva, T., Gatti, D. L., Rosen, B. P., and Edwards, B. F. P.: Insights into the structure, solvation, and mechanism of ArsC arsenate reductase, a novel arsenic detoxification enzyme, Structure, 9, 1071–1081, https://doi.org/10.1016/s0969-2126(01)00672-4, 2001.
Maupin-Furlow, J. A., Rosentel, J. K., Lee, J. H., Deppenmeier, U., Gunsalus, R., and Shanmugam, K.: Genetic analysis of the modABCD (molybdate transport) operon of Escherichia coli, J. Bacteriol., 177, 4851–4856, https://doi.org/10.1128/jb.177.17.4851-4856.1995, 1995.
Mengoni, A. and Bazzicalupo, M.: The statistical treatment of data and the Analysis of MOlecular VAriance (AMOVA) in molecular microbial ecology, Ann. Microbiol., 52, 95–102, 2002.
Newton, G. L., Av-Gay, Y., and Fahey, R. C.: A novel mycothiol-dependent detoxification pathway in Mycobacteria involving mycothiol S-Conjugate amidase, Biochemistry, 39, 10739–10746, https://doi.org/10.1021/bi000356n, 2000.
Newton, G. L., Buchmeier, N., and Fahey, R. C.: Biosynthesis and Functions of Mycothiol, the Unique Protective Thiol of Actinobacteria, Microbiol. Mol. Biol. Rev., 72, 471–494, https://doi.org/10.1128/MMBR.00008-08, 2008.
Nies, D. H.: Efflux-mediated heavy metal resistance in prokaryotes, FEMS Microbiol. Rev., 27, 313–339, https://doi.org/10.1016/S0168-6445(03)00048-2, 2003.
Novoa-Aponte, L., Ramírez, D., and Argüello, J. M.: The interplay of the metallosensor CueR with two distinct CopZ chaperones defines copper homeostasis in Pseudomonas aeruginosa, J. Biol. Chem., 294, 4934–4945, https://doi.org/10.1074/jbc.RA118.006316, 2019.
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A.: metaSPAdes: a new versatile metagenomic assembler, Genome Res., 27, 824–834, https://doi.org/10.1101/gr.213959.116, 2017.
Osman, D., Waldron, K. J., Denton, H., Taylor, C. M., Grant, A. J., Mastroeni, P., Robinson, N. J., and Cavet, J. S.: Copper homeostasis in Salmonella is atypical and copper-CueP is a major periplasmic metal complex, J. Biol. Chem., 285, 25259–25268, https://doi.org/10.1074/jbc.m110.145953, 2010.
Outten, F. W., Huffman, D. L., Hale, J. A., and O'Halloran, T. V.: The independent cue and cus systems confer copper tolerance during aerobic and anaerobic growth in Escherichia coli, J. Biol. Chem., 276, 30670–30677, https://doi.org/10.1074/jbc.m104122200, 2001.
Papp-Wallace, K. M. and Maguire, M. E.: Manganese transport and the role of manganese in virulence, Annu. Rev. Microbiol., 60, 187–209, https://doi.org/10.1146/annurev.micro.60.080805.142149, 2006.
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W.: CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes, Genome Res., 25, 1043–1055, https://doi.org/10.1101/gr.186072.114, 2015.
Parks, D. H., Chuvochina, M., Chaumeil, P.-A., Rinke, C., Mussig, A. J., and Hugenholtz, P.: Selection of representative genomes for 24,706 bacterial and archaeal species clusters provide a complete genome-based taxonomy, BioRxiv [preprint], https://doi.org/10.1101/771964, 2019.
Paul Chowdhury, S., Babin, D., Sandmann, M., Jacquiod, S., Sommermann, L., Sørensen, S. J., Fliessbach, A., Mäder, P., Geistlinger, J., and Smalla, K.: Effect of long-term organic and mineral fertilization strategies on rhizosphere microbiota assemblage and performance of lettuce, Environ. Microbiol., 21, 2426–2439, https://doi.org/10.1111/1462-2920.14631, 2019.
Rademacher, C. and Masepohl, B.: Copper-responsive gene regulation in bacteria, Microbiology, 158, 2451, https://doi.org/10.1099/mic.0.058487-0, 2012.
Rae, T. D., Schmidt, P. J., Pufahl, R. A., Culotta, V. C., and O'Halloran, T. V.: Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase, Science, 284, 805–808, https://doi.org/10.1126/science.284.5415.805, 1999.
Redinbo, M. R., Yeates, T. O., and Merchant, S.: Plastocyanin: structural and functional analysis, J. Bioenerg. Biomembr., 26, 49–66, https://doi.org/10.1007/BF00763219, 1994.
Rensing, C. and Mitra, B.: Zinc, cadmium, and lead resistance and homeostasis, in: Molecular microbiology of heavy metals, Springer, 321–341, https://doi.org/10.1007/7171_2006_083, 2007.
Richard, H. and Foster, J. W.: Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential, J. Bacteriol., 186, 6032–6041, https://doi.org/10.1128/JB.186.18.6032-6041.2004, 2004.
Ridge, J. P., Lin, M., Larsen, E. I., Fegan, M., McEwan, A. G., and Sly, L. I.: A multicopper oxidase is essential for manganese oxidation and laccase-like activity in Pedomicrobium sp. ACM 3067, Environ. Microbiol., 9, 944–953, https://doi.org/10.1111/j.1462-2920.2006.01216.x, 2007.
Rosen, B. P.: Biochemistry of arsenic detoxification, FEBS Lett., 529, 86–92, https://doi.org/10.1016/S0014-5793(02)03186-1, 2002.
Rowland, J. L. and Niederweis, M.: A multicopper oxidase is required for copper resistance in Mycobacterium tuberculosis, J. Bacteriol., 195, 3724–3733, https://doi.org/10.1128/jb.00546-13, 2013.
Sambrook, J. and Russell, D. W.: Molecular cloning: A laboratory manual, 3rd Edn., Cold spring harbor laboratory press, Cold Spring Harbor, New York, ISBN 978-0-87969-577-4, 2001.
Schippers, A. and Sand, W.: Bacterial leaching of metal sulfides proceeds by two indirect mechanisms via thiosulfate or via polysulfides and sulfur, Appl. Environ. Microbiol., 65, 319–321, https://doi.org/10.1128/AEM.65.1.319-321.1999, 1999.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. J., Sahl, J. W., Stres, B., Thallinger, G. G., Van Horn, D. J., and Weber, C. F.: Introducing mothur: Open-Source, platform-independent, community-supported software for describing and comparing microbial communities, Appl. Environ. Microbiol., 75, 7537, https://doi.org/10.1128/AEM.01541-09, 2009.
Schmidt, A., Haferburg, G., Sineriz, M., Merten, D., Büchel, G., and Kothe, E.: Heavy metal resistance mechanisms in actinobacteria for survival in AMD contaminated soils, Geochemistry, 65, 131–144, https://doi.org/10.1016/j.chemer.2005.06.006, 2005.
Seemann, T.: Prokka: rapid prokaryotic genome annotation, Bioinformatics, 30, 2068–2069, https://doi.org/10.1093/bioinformatics/btu153, 2014.
Self, W. T., Grunden, A. M., Hasona, A., and Shanmugam, K. T.: Molybdate transport, Res. Microbiol., 152, 311–321, https://doi.org/10.1007/s002030050508, 2001.
Shi, X., Festa, R. A., Ioerger, T. R., Butler-Wu, S., Sacchettini, J. C., Darwin, K. H., and Samanovic, M. I.: The copper-responsive RicR regulon contributes to Mycobacterium tuberculosis virulence, MBio, 5, e00876-13, https://doi.org/10.1128/mBio.00876-13, 2014.
Siegmund, H., Trappe, J., and Oschmann, W.: Sequence stratigraphic and genetic aspects of the Tournaisian “Liegender Alaunschiefer” and adjacent beds, Int. J. Earth Sci., 91, 934–949, https://doi.org/10.1007/s00531-001-0252-9, 2002.
Silver, S. and Hobman, J. L.: Mercury microbiology: resistance systems, environmental aspects, methylation, and human health, in: Molecular Microbiology of Heavy Metals, edited by: Nies, D. H., and Silver, S., Springer Berlin Heidelberg, Berlin, Heidelberg, 357–370, https://doi.org/10.1007/7171_2006_085, 2007.
Silver, S. and Phung, L. T.: A bacterial view of the periodic table: genes and proteins for toxic inorganic ions, J. Ind. Microbiol. Biotechnol., 32, 587–605, https://doi.org/10.1007/s10295-005-0019-6, 2005.
Silver, S. and Walderhaug, M.: Gene regulation of plasmid-and chromosome-determined inorganic ion transport in bacteria, Microbiol. Mol. Biol. R., 56, 195–228, https://doi.org/10.1128/mr.56.1.195-228.1992, 1992.
Smaldone, G. T. and Helmann, J. D.: CsoR regulates the copper efflux operon copZA in Bacillus subtilis, Microbiology, 153, 4123, https://doi.org/10.1099/mic.0.2007/011742-0, 2007.
Stecher, G., Tamura, K., and Kumar, S.: Molecular evolutionary genetics analysis (MEGA) for macOS, Mol. Biol. Evol., 37, 1237–1239, https://doi.org/10.1093/molbev/msz312, 2020.
Stribrny, B.: Die Kupfererzlagerstätte Marsberg im Rheinischen Schiefergebirge – Rückblick und Stand der Forschung, Erzmetall, 40, 423–427, 1987.
Stribrny, B. and Urban, H.: Zur Geologie und Lagerstättenbildung des Kupfererzvorkommens von Marsberg im Rheinischen Schiefergebirge, On the geology and formation of the copper ore deposit of Marsberg in the Rhenish region Slate Mountains, 347–416, https://nwbib.de/HT013094414 (last access: 2 April 2021), 2000.
Thomas Iv, J. C., Oladeinde, A., Kieran, T. J., Finger Jr, J. W., Bayona-Vásquez, N. J., Cartee, J. C., Beasley, J. C., Seaman, J. C., McArthur, J. V., and Rhodes Jr., O. E.: Co-occurrence of antibiotic, biocide, and heavy metal resistance genes in bacteria from metal and radionuclide contaminated soils at the Savannah River Site, Microb. Biotechnol., 13, 1179–1200, https://doi.org/10.1111/1751-7915.13578, 2020.
Thompson, A. K., Gray, J., Liu, A., and Hosler, J. P.: The roles of Rhodobacter sphaeroides copper chaperones PCuAC and Sco (PrrC) in the assembly of the copper centers of the aa3-type and the cbb3-type cytochrome c oxidases, Biochim. Biophys. Acta, 1817, 955–964, https://doi.org/10.1016/j.bbabio.2012.01.003, 2012.
Tiwari, K. and Gupta, R. K.: Rare actinomycetes: a potential storehouse for novel antibiotics, Cr. Rev. Biotechn., 32, 108–132, https://doi.org/10.3109/07388551.2011.562482, 2012.
Tsai, M.-F. and Miller, C.: Substrate selectivity in arginine-dependent acid resistance in enteric bacteria, P. Natl. Acad. Sci. USA, 110, 5893, https://doi.org/10.1073/pnas.1301442110, 2013.
Urban, H., Stribrny, B., Zereini, F., and Ye, Y.: Geochemistry and metallogenesis of Lower Carboniferous black shale-hosted ore deposits, NE Rhenish Massif, FR Germany, Ore Geol. Rev., 9, 427–443, https://doi.org/10.1016/0169-1368(94)00023-H, 1995.
Villafane, A., Voskoboynik, Y., Ruhl, I., Sannino, D., Maezato, Y., Blum, P., and Bini, E.: CopR of Sulfolobus solfataricus represents a novel class of archaeal-specific copper-responsive activators of transcription, Microbiology, 157, 2808, https://doi.org/10.1099/mic.0.051862-0, 2011.
Vita, N., Landolfi, G., Baslé, A., Platsaki, S., Lee, J., Waldron, K. J., and Dennison, C.: Bacterial cytosolic proteins with a high capacity for Cu(I) that protect against copper toxicity, Sci. Rep., 6, 39065, https://doi.org/10.1038/srep39065, 2016.
Waters, L. S., Sandoval, M., and Storz, G.: The Escherichia coli MntR miniregulon includes genes encoding a small protein and an efflux pump required for manganese homeostasis, J. Bacteriol., 193, 5887–5897, https://doi.org/10.1128/jb.05872-11, 2011.
Wegner, C.-E. and Liesack, W.: Unexpected dominance of elusive Acidobacteria in early industrial soft coal slags, Front. Microbiol., 8, 1023, https://doi.org/10.3389/fmicb.2017.01023, 2017.
Wolfram, L., Friedrich, B., and Eitinger, T.: The Alcaligenes eutrophus protein HoxN mediates nickel transport in Escherichia coli, J. Bacteriol., 177, 1840–1843, https://doi.org/10.1128/jb.177.7.1840-1843.1995, 1995.
Wu, Y.-W., Simmons, B. A., and Singer, S. W.: MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets, Bioinformatics, 32, 605–607, https://doi.org/10.1093/bioinformatics/btv638, 2015.
Xia, Z., Lei, L., Zhang, H.-Y., and Wei, H.-L.: Characterization of the ModABC molybdate transport system of Pseudomonas putida in nicotine degradation, Front. Microbiol., 9, 3213, https://doi.org/10.3389/fmicb.2018.03030, 2018.
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., Zhang, G., Gu, Y. Q., Coleman-Derr, D., and Xia, Q.: OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species, Nucl. Acid. Res., 47, W52–W58, https://doi.org/10.1093/nar/gkz333, 2019.
Yabe, S., Aiba, Y., Sakai, Y., Hazaka, M., and Yokota, A.: Thermogemmatispora onikobensis gen. nov., sp. nov. and Thermogemmatispora foliorum sp. nov., isolated from fallen leaves on geothermal soils, and description of Thermogemmatisporaceae fam. nov. and Thermogemmatisporales ord. nov. within the class Ktedonobacteria, Int. J. Syst. Evol. Micr., 61, 903–910, https://doi.org/10.1099/ijs.0.024877-0, 2011.
Yabe, S., Sakai, Y., Abe, K., and Yokota, A.: Diversity of Ktedonobacteria with Actinomycetes-like Morphology in terrestrial environments, Microb. Environ., 32, 61–70, https://doi.org/10.1264/jsme2.ME16144, 2017a.
Yabe, S., Sakai, Y., Abe, K., Yokota, A., Také, A., Matsumoto, A., Sugiharto, A., Susilowati, D., Hamada, M., and Nara, K.: Dictyobacter aurantiacus gen. nov., sp. nov., a member of the family Ktedonobacteraceae, isolated from soil, and emended description of the genus Thermosporothrix, Int. J. Syst. Evol. Micr., 67, 2615–2621, https://doi.org/10.1099/ijsem.0.001985, 2017b.
Zaballa, M.-E., Abriata, L. A., Donaire, A., and Vila, A. J.: Flexibility of the metal-binding region in apo-cupredoxins, P. Natl. Acad. Sci. USA, 109, 9254–9259, https://doi.org/10.1073/pnas.1119460109, 2012.
Zeng, X.-C., Yang, Y., Shi, W., Peng, Z., Chen, X., Zhu, X., and Wang, Y.: Microbially mediated methylation of arsenic in the arsenic-rich soils and sediments of Jianghan Plain, Front. Microbiol., 9, 1389, https://doi.org/10.3389/fmicb.2018.01389, 2018.
Zhang, Z., Zhang, Z., Chen, H., Liu, J., Liu, C., Ni, H., Zhao, C., Ali, M., Liu, F., and Li, L.: Surface Mn (II) oxidation actuated by a multicopper oxidase in a soil bacterium leads to the formation of manganese oxide minerals, Sci. Rep., 5, 10895, https://doi.org/10.1038/srep10895, 2015.