the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Dec 2022
| 14 Dec 2022
Depth-related patterns in microbial community responses to complex organic matter in the western North Atlantic Ocean
John Paul Balmonte
Adrienne Hoarfrost
Sherif Ghobrial
Carol Arnosti
Oceanic bacterial communities process a major fraction of marine organic carbon. A substantial portion of this carbon transformation occurs in the mesopelagic zone, and a further fraction fuels bacteria in the bathypelagic zone. However, the capabilities and limitations of the diverse microbial communities at these depths to degrade high-molecular-weight (HMW) organic matter are not well constrained. Here, we compared the responses of distinct microbial communities from North Atlantic epipelagic (0–200 m), mesopelagic (200–1000 m), and bathypelagic (1000–4000 m) waters at two open-ocean stations to the same input of diatom-derived HMW particulate and dissolved organic matter. Microbial community composition and functional responses to the input of HMW organic matter – as measured by polysaccharide hydrolase, glucosidase, and peptidase activities – were very similar between the stations, which were separated by 1370 km but showed distinct patterns with depth. Changes in microbial community composition coincided with changes in enzymatic activities: as bacterial community composition changed in response to the addition of HMW organic matter, the rate and spectrum of enzymatic activities increased. In epipelagic mesocosms, the spectrum of peptidase activities became especially broad and glucosidase activities were very high, a pattern not seen at other depths, which, in contrast, were dominated by leucine aminopeptidase and had much lower peptidase and glucosidase rates in general. The spectrum of polysaccharide hydrolase activities was enhanced particularly in epipelagic and mesopelagic mesocosms, with fewer enhancements in rates or spectrum in bathypelagic waters. The timing and magnitude of these distinct functional responses to the same HMW organic matter varied with depth. Our results highlight the importance of residence times at specific depths in determining the nature and quantity of organic matter reaching the deep sea.
- Article
(1581 KB) - Full-text XML
-
Supplement
(3355 KB) - BibTeX
- EndNote





Heterotrophic microbial communities play a key role in global biogeochemical cycles by processing up to 50 % of the primary productivity produced by phytoplankton in the ocean (Azam, 1998). Much of this organic matter is quickly consumed, transformed, and remineralized in the upper ocean, while most of the remainder is consumed in the mesopelagic zone (200–1000 m), with only a fraction reaching deeper depths (Wakeham et al., 1997; Benner and Amon, 2015). The organic matter reaching the bathypelagic zone (1000–4000 m) typically contains low proportions of chemically characterizable carbohydrates, amino acids, and lipids (Benner and Amon, 2015). However, recent investigations have documented rapid transport of freshly produced organic matter from the upper ocean to the bathypelagic via fast-sinking particles, thus injecting fresh epipelagic-derived organic matter into the deep (Mestre et al., 2018; Ruiz-González et al., 2020; Poff et al., 2021). The extent to which this organic matter is transformed and remineralized once reaching meso- and bathypelagic zones is ultimately determined by the metabolic capabilities of the heterotrophic microbial communities present at these depths.
A key step in microbial remineralization of complex high-molecular-weight (HMW) particulate and dissolved organic matter is its initial hydrolysis to pieces small enough for cellular uptake (Weiss et al., 1991; Arnosti, 2011). Proteins and polysaccharides, which account for the majority of HMW organic matter and are preferentially utilized by bacteria (Amon et al., 2001; Hedges et al., 2001), require distinct, structurally specific extracellular enzymes in order for hydrolysis to occur. The specific capabilities and limitations of natural microbial communities, particularly those in the meso- and bathypelagic zones, to enzymatically hydrolyze this HMW organic matter are not well-defined, however. Previous studies examining the response of microbial communities to organic matter addition have typically focused on epipelagic water communities, often using a mixture of both low- and high-molecular-weight material (e.g., Beier et al., 2015; Luria et al., 2017) to examine community responses. Moreover, the studies that have measured the enzymatic responses of deep-ocean communities have relied for the most part on a few small substrate proxies to assess exo-acting (terminal-unit cleaving) enzyme activities (e.g., Baltar et al., 2010; Sebastián et al., 2021); these substrate proxies do not yield information about the endo-acting (mid-chain cleaving) enzymes essential for degradation of HMW organic matter.
As an alternative, the activities of endo-acting enzymes can be measured using fluorescently labeled polysaccharides (Arnosti, 2003) that can probe structure-related differences in polysaccharide hydrolase activities. Such studies have revealed considerable differences in the rate and spectrum of enzyme activities with location and depth in the ocean, with the spectrum of substrates hydrolyzed typically decreasing with depth (Steen et al., 2012; Hoarfrost and Arnosti, 2017; Balmonte et al., 2018). Distinct spatial- and depth-related patterns in endopeptidase activities have also been found to exist (Balmonte et al., 2021). These patterns coincide with patterns in the depth stratification of microbial taxa, genes, and metabolic potential (DeLong et al., 2006; Sunagawa et al., 2015; Guerrero-Feijóo et al., 2016). Together, these results suggest that microbial communities at different depths and locations vary considerably in their abilities to initiate the remineralization of HMW organic matter.
As an initial investigation of the enzymatic response of spatially distinct microbial communities to complex organic matter, we previously added moderate quantities of HMW particulate and dissolved organic matter to epipelagic water and bottom water collected at a shelf station and at an offshore station in the western North Atlantic Ocean. Epipelagic and bottom water communities from a shelf and an open-ocean station rapidly responded to HMW organic matter addition, with greater enhancements of rates and a broader spectrum of enzyme activities in communities from the shelf and from epipelagic water relative to the offshore bottom water (Balmonte et al., 2019). Nonetheless, a distinct enhancement in enzyme activities measurable in bathypelagic water from a depth of 4594 m demonstrated that an active heterotrophic community could respond in comparatively short order to an input of HMW organic matter. This initial assessment showed that microbial communities at a shelf and an offshore station responded to an addition of complex HMW organic matter, but the nature of that enzymatic response differed in some key respects. Epipelagic communities, both on the shelf and offshore, responded with higher rates and a broader spectrum of enzymatic activities, while the bottom water community offshore did not exhibit the same breadth of enzymatic activities. Moreover, the bottom water community took longer to respond to the addition of HMW organic matter and exhibited lower rates of enzymatic hydrolysis than the epipelagic and the shelf bottom water communities.
In order to better define the potential and possible enzymatic limitations of open-ocean microbial communities, in the present study we sought to determine with greater resolution the depth gradients in community function and composition and to determine the extent to which our previous (single-station) open-ocean results apply across larger spatial gradients. We therefore selected two open-ocean stations 1370 km apart, where we investigated organic matter transformation processes in the mesopelagic ocean, where substantial amounts of the organic matter sinking from the epipelagic is remineralized (Wakeham et al., 1997; Benner and Amon, 2015), and compared these results with transformation processes in the epipelagic and bathypelagic ocean. We added moderate quantities (658 µM) of particulate and HMW dissolved organic matter derived from Thalassiosira weissflogii, a widespread, abundant diatom (Hartley et al., 1996), to triplicate mesocosms from each depth and tracked microbial community responses and enzyme activities associated with metabolism of two major classes of marine organic matter, polysaccharides and proteins. By carrying out same experiments at two open-ocean stations, one in the Gulf Stream and one in the Sargasso Sea, we investigated the extent to which spatially separated, depth-stratified microbial communities may be functionally redundant – or not – in terms of their abilities to hydrolyze the same complex organic matter.
2.1 Sample sites and sample collection
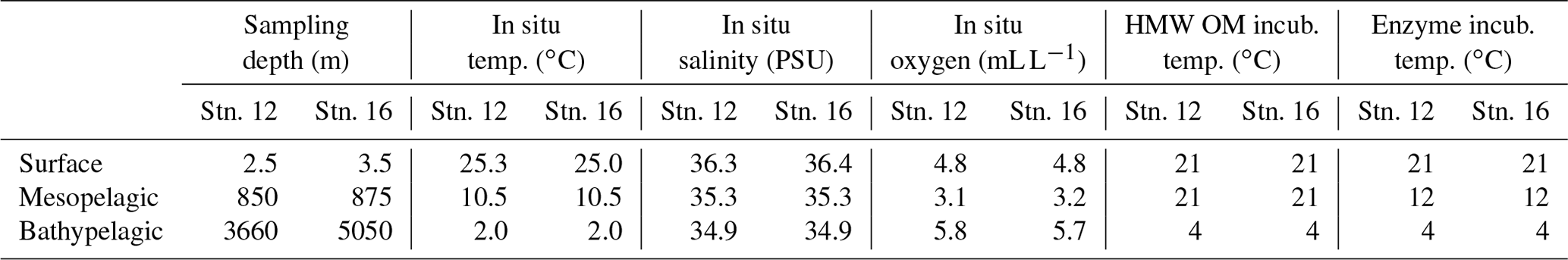
Water was collected aboard the R/V Endeavor in July 2016 at two locations in the western North Atlantic Ocean: stn. 12 (station; 36∘0′12.12′′ N, 73∘8′38.46′′ W), with surface waters in the Gulf Stream, and stn. 16 (35∘59′43.20′′ N, 58∘2′40.80′′ W), located in the North Atlantic Gyre (Fig. S1 in the Supplement). We focused on epipelagic, mesopelagic (at the oxygen minimum zone, identified from the conductivity–temperature–depth profile; CTD), and bathypelagic water (bottom water), corresponding to depths of 2.5, 850, and 3660 m at stn. 12 and 3.5, 875, and 5050 m at stn. 16, respectively. Water was transferred from the Niskin bottles to 20 L carboys (acid-washed and rinsed, then rinsed again with sample water prior to filling). Each carboy was filled from a separate Niskin bottle for biological replicates. Triplicate carboys from each depth were amended with moderate (25 mg L−1) quantities of material isolated from Thalassiosira weissflogii, corresponding to approximately 658 µM of HMW particulate + dissolved organic carbon (see below and Balmonte et al., 2019). One unamended carboy from each depth served as an incubation control. Carboys were stored in the dark at in situ or near in situ temperatures: samples from the epipelagic and mesopelagic were incubated at 21 ∘C, and bottom water samples were incubated at 4 ∘C (Table 1). At each subsampling time point (0, 2, 7, and 16 d after the addition of HMW substrate), the carboys were mixed, and subsamples for measurements of cell counts, bacterial production, peptidase and glucosidase activities, and bacterial community composition were collected. Incubations to measure polysaccharide hydrolase activities were initiated at the 16 d time point (see below).
Table 1Sampling depth, in situ characteristics, and incubation temperatures of water collected for mesocosms at each station and depth. OM: organic matter, PSU: practical salinity unit.
2.2 High-molecular-weight organic matter preparation
A HMW substrate was prepared from the diatom Thalassiosira weissflogii (Instant Algae, Reed Mariculture), as described in Balmonte et al. (2019). In brief, frozen and thawed cells were homogenized with a tissue grinder and dialyzed in a 10 kD membrane (SpectraPor), and the retentate (HMW dissolved organic matter plus particulate organic matter) was lyophilized, autoclaved, and lyophilized again. The final HMW Thalassiosira weissflogii substrate had a total carbohydrate concentration of 6.15 % and a C : N ratio of 6 : 1 (Balmonte et al., 2019).
2.3 Bacterial productivity
Bacterial productivity was measured aboard ship according to the methods of Kirchman et al. (1985) and Kirchman (2001). Samples were incubated in the dark at in situ temperatures between 12 and 24 h. Bacterial protein production was calculated from leucine incorporation rates using the equation of Simon and Azam (1989), and bacterial carbon production was determined by multiplying bacterial protein production by 0.86 (Simon and Azam, 1989; Kirchman, 2001).
2.4 Enzymatic hydrolysis measurements
Peptidase and glucosidase activities were measured immediately after the addition of HMW organic matter to the amended carboys, as well as 2, 7, and 16 d following the amendment. These activities were measured using small substrates (glucose, leucine, or peptides) labeled with 4-methyl-coumaryl-7-amide (MCA) or methylumbelliferone (MUF). Exo-acting (terminal-unit cleaving) glucosidase activities were measured with MUF-α- and MUF-β-glucose, and leucine-MCA was used to measure exopeptidase activities, after the approach of Hoppe (1983). MUF-α- and MUF-β-glucose measure the activities of α- and β-glucosidases, respectively, which are exo-acting enzymes that hydrolyze terminal glucose units that are initially linked to other molecules in an α or β orientation, respectively. Endopeptidase activities were measured with Boc-Gln-Ala-Arg-MCA (QAR, using one-letter amino acid abbreviations) and N-t-Boc-Phe-Ser-Arg-MCA (FSR) for trypsin activities, while Ala-Ala-Phe-MCA (AAF) and N-succinyl-Ala-Ala-Pro-Phe-MCA (AAPF) were used to measure chymotrypsin activities. Activities were measured in triplicate using a plate reader, following Balmonte et al. (2019), using substrate concentrations of 150 µM, a concentration based on substrate saturation curves of leucine aminopeptidase and β-glucosidase in stn. 9 epipelagic waters (76∘36′6.12′′ N, 34∘36′6.552′′ W). The fluorescence of autoclaved seawater with substrate (controls), as well as live seawater with no substrate (blanks), was also measured. Samples were incubated close to in situ temperature and measured at multiple time points – at 0, 6, 12, 18, 24, 36, and 48 h. Fluorescence readings were converted to activities using a standard curve of free fluorophores (MCA, MUF) in seawater. Hydrolysis rates were averaged over the first 48 h of measurements for each sampling day.
Polysaccharide hydrolase activities were measured using six fluorescently labeled polysaccharides (pullulan, laminarin, xylan, fucoidan, arabinogalactan, and chondroitin sulfate; Arnosti, 2003), chosen for their varying structural complexities and abundances in the ocean. For example, laminarin, a storage glucan found in phytoplankton, including diatoms, is highly abundant in the ocean (Alderkamp et al., 2007; Becker et al., 2020; Vidal-Melgosa et al., 2021) and plays a substantial role in oceanic carbon cycling as it is rapidly hydrolyzed by marine bacteria (e.g., Arnosti et al., 2011; Hoarfrost and Arnosti, 2017; Balmonte et al., 2021). More complex polysaccharides, including arabinogalactan, a polysaccharide found in green microalgae, and fucoidan, a cell wall polysaccharide produced by brown algae that is also present in diatom exudates (Vidal-Melgosa et al., 2021), are less readily degraded by marine bacteria and may persist in the ocean for longer periods of time (Sichert et al., 2020; Vidal-Melgosa et al., 2021). Additionally, marine bacteria capable of degrading the chosen polysaccharides have been identified, including bacteria in the classes Gammaproteobacteria and Bacteroidia that consume laminarin (Alderkamp et al., 2007; Teeling et al., 2012; Vidal-Melgosa et al., 2021), while Verrucomicrobia are capable of fucoidan hydrolysis (Sichert et al., 2020). The other polysaccharides (pullulan, xylan, and chondroitin sulfate) are known to be marine-derived, and/or their hydrolysis of these substrates is carried out by marine bacteria (e.g., Arnosti and Repeta, 1994; Wegner et al., 2013; Araki et al., 2000).
Measurements of these six polysaccharides were initiated in water from the amended and unamended mesocosms 16 d after the start of the experiment, an initial time lag that should allow for the microbial community in amended mesocosms to respond – via enzyme expression as well as shifts in community composition – to the addition of Thalassiosira-derived material. Incubations were carried out after Balmonte et al. (2019). All incubations were kept in the dark at in situ temperatures. Subsamples were collected 0, 2, 5, 10, 17, and 30 d after polysaccharide addition. Hydrolysis rates were calculated as previously described (Arnosti, 2003). Note that all enzymatic hydrolysis rates – polysaccharide hydrolase as well as peptidase – should be considered potential rates, since added substrate is in competition with naturally occurring substrates for enzyme active sites.
2.5 16S rRNA sequencing and phylogenetic analysis
To analyze bacterial community composition, 250 to 2500 mL of seawater from each carboy was filtered using a vacuum pump through a 0.2 µm pore size, 47 mm diameter Whatman Nuclepore Track-Etched Membrane filter (see Table S1 in the Supplement for volumes filtered). Samples were stored at −80 ∘C until analysis. At least one-quarter of each filter was cut with a sterile razor blade and used for DNA extraction. Analysis of sample duplicates (i.e., pieces of different filters from the same sample) and/or filter duplicates (duplicate quarters of a single filter) were also analyzed for a select number of samples. DNA was extracted using a DNeasy PowerSoil Kit (Qiagen) according to manufacturer protocol. The 16S rRNA gene was sequenced at the University of North Carolina at Chapel Hill (UNC) Core Microbiome Facility with Illumina MiSeq PE 2×250. Amplification of the hypervariable regions V1 and V2 of the 16s rRNA gene was conducted using the 8F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and 338R (5′-GC TGC CTC CCG TAG GAG T-3′) primers with the Illumina-specific forward primer overhang adapter (5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG-3′) and reverse overhang adapter (5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G-3′).
Sequences were imported into QIIME2 for analysis (version 2017.12; https://qiime2.org, last access: 25 January 2018; Bolyen et al., 2019), where, after demultiplexing, primers were trimmed using cutadapt (Martin, 2011) and denoising, dereplicating, filtering of chimeras and singletons, and merging of paired end reads was conducted using DADA2 (Callahan et al., 2016). OTUs (operational taxonomic units) were clustered and picked de novo at a sequence similarity of 97 % using vsearch (Rognes et al., 2016). Taxonomy was assigned using a naïve Bayes classifier that was trained using reference sequences with the 8F and 338R primers from the SILVA database (version 128; Pruesse et al., 2007). Chloroplasts were removed, and samples were rarefied to an even sampling depth of 10 810 sequences using the phyloseq package in R (version 1.19.1; McMurdie and Holmes, 2013). Raw sequence files can be accessed on the National Center for Biotechnology Information (NCBI) Sequence Read Archive under the accession number PRJNA480640.
2.6 Statistical analyses
To test for differences in bacterial productivity, bacterial abundance, polysaccharide hydrolase activities, and peptidase and glucosidase activities between different stations, depths, and treatments over time, ANOVA was performed using the nlme package (version 3.1-131; Pinheiro et al., 2018) in R (R Core Team, 2017). Bacterial abundance, richness, and evenness were log-transformed, while bacterial productivity and all enzymatic activities were transformed according to ln (x+0.5), in order to meet the assumptions of an ANOVA. In each test, the station, depth, treatment, and time point were considered fixed variables, and the mesocosm was considered a random variable. According to ANOVAs, the four-way interaction (between the station, depth, treatment, and time point) was not significant for bacterial productivity, bacterial abundance, or any of the enzymatic activities, and these were therefore excluded from the ANOVA test.
Differences in bacterial community composition were visualized using non-metric multidimensional scaling (NMDS) based on the Bray–Curtis dissimilarity index using the phyloseq package in R (version 1.19.1; McMurdie and Holmes, 2013). To test for differences in bacterial community composition between groups, PERMANOVAs (permutational multivariate ANOVAs) were conducted using the adonis function (vegan) (version 2.4-6; Oksanen et al., 2018) in R. Estimates of richness and evenness of bacterial community composition were calculated in R using the estimate richness function in the phyloseq package (version 1.19.1; McMurdie and Holmes, 2013). To examine the correlation between enzymatic activities and bacterial community composition, Mantel tests using the Pearson correlation method were conducted on the Bray–Curtis dissimilarity index of bacterial community composition and a Euclidean distance matrix computed from peptidase and glucosidase activities using the vegan package in R (version 2.4-6; Oksanen et al., 2018).
3.1 Water mass characteristics
The two stations in the western North Atlantic Ocean, separated by a distance of 1370 km, included the same water masses at each depth, based on temperature and salinity characteristics: epipelagic water at both stations was North Atlantic Surface Water, water from the mesopelagic zone (850 m at stn. 12; 875 m at stn. 16) was North Atlantic Central Water, and bathypelagic water at both stations was North Atlantic Deep Water (Talley, 2011; Heiderich and Todd, 2020; Table 1; Figs. S1, S2 in the Supplement). Water mass characteristics in the epi-, meso-, and bathypelagic zones were substantially different from one another (see Table 1; Fig. S2), enabling the examination of bacterial communities experiencing distinct environmental conditions at two open-ocean stations – one situated within the Gulf Stream and one located in the Sargasso Sea.
3.2 Peptidase and glucosidase activities
Peptidases hydrolyze peptides and proteins to smaller substrates, whereas glucosidases hydrolyze terminal glucose units from larger molecules. Since peptidase and glucosidase activities differed by depth but not by station in both amended and unamended mesocosms (ANOVA, p=0.2424), in the following sections, data from the same depths at the two stations are presented together, yielding two unamended and six amended mesocosms per depth. Data from individual mesocosms are presented in the figures in the Supplement.
In unamended epipelagic water mesocosms, all seven peptidase and glucosidase substrates were hydrolyzed at rates higher than in the mesopelagic or bathypelagic waters (Figs. 1; S3); in epipelagic waters, endopeptidase activities (i.e., AAF-chymotrypsin, AAPF-chymotrypsin, QAR-trypsin, and FSR-trypsin) were also a higher fraction of summed activity than at other depths (Fig. S3). Bathypelagic waters were dominated by leucine-MCA and AAF-chymotrypsin activities; no AAPF-chymotrypsin activity was detected (Figs. 1; S3).
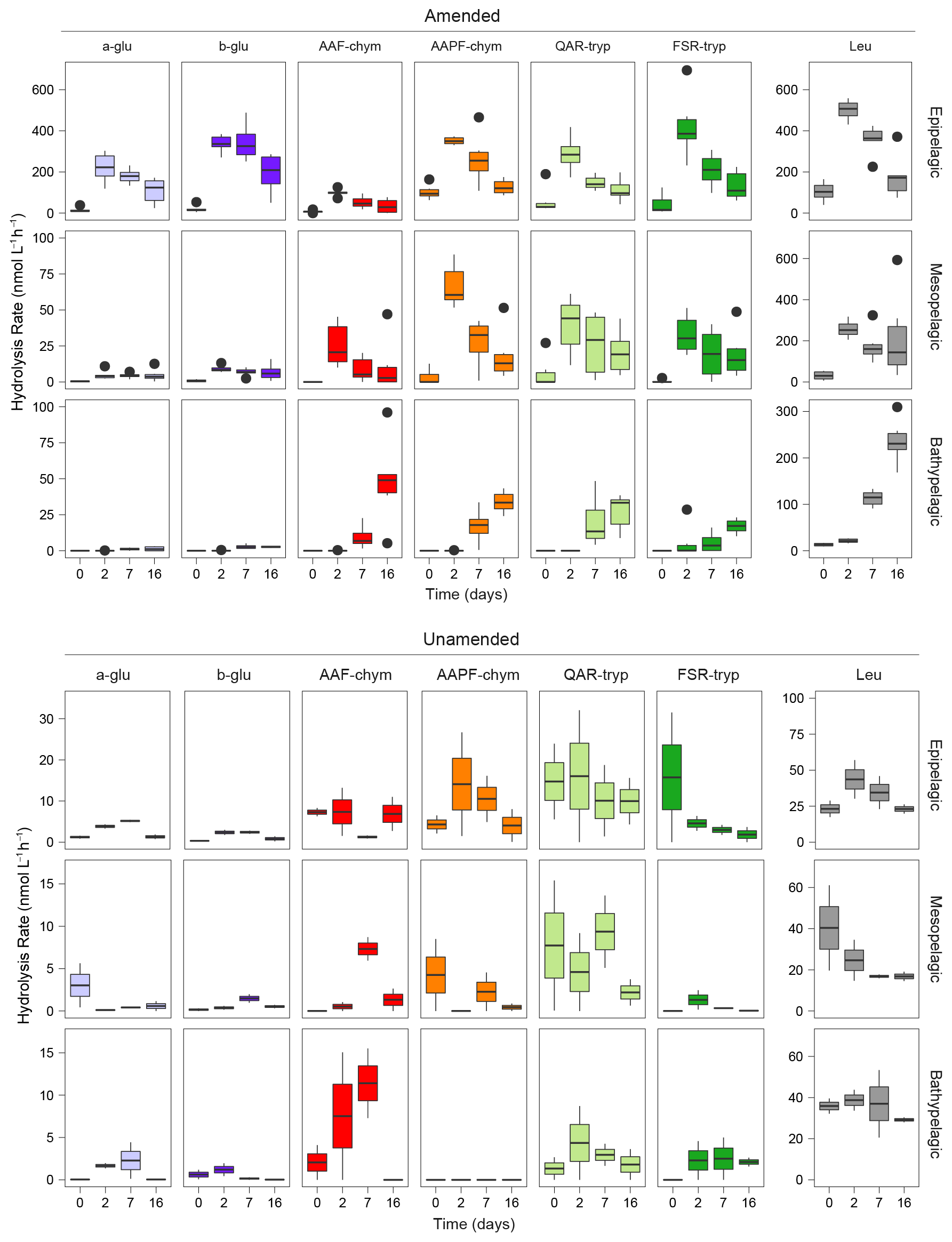
Figure 1Peptidase and glucosidase activities in amended and unamended mesocosms 0, 2, 7, and 16 d after the addition of HMW organic matter to amended mesocosms. Rates are an average between the two stations. Note the difference in scales between the amended and unamended mesocosms. a-glu: α-glucose, b-glu: β-glucose, AAF-chym: AAF (chymotrypsin), AAPF-chym: AAPF (chymotrypsin), QAR-tryp: QAR (trypsin), FSR-tryp: FSR (trypsin), and Leu: leucine-MCA.
Addition of HMW organic matter led to a substantial increase in the rates and spectrum of peptidase and glucosidase activities (Fig. 1) (ANOVA, p<0.0001). The responses of the six amended mesocosms per depth, drawn from six different Niskin bottles across two different stations, were very similar (Fig. S3). The timing of these responses to added organic matter varied with depth, however, suggesting that depth had a stronger influence on enzymatic responses than location. At t0, immediately after HMW organic matter addition, hydrolysis rates and patterns in amended mesocosms were similar to the unamended mesocosms. By the 2 d time point, activities had increased by approximately an order of magnitude in the epipelagic mesocosms, and the hydrolysis rates of the substrates became more even. In amended mesopelagic mesocosms, after 2 d all activities were approximately a factor of 5 greater than in unamended mesocosms. However, a shift in enzymatic response to HMW organic matter amendment, defined as the point at which the rates and spectrum of peptidase and glucosidase activities increased significantly, in bathypelagic mesocosms was not observed until 7 d following the addition (Figs. 1; S3).
The stimulation of specific peptidase and glucosidase activities in amended mesocosms also differed by depth (Fig. 1) (ANOVA, p<0.0001), pointing to further depth-related functional capabilities between epipelagic, mesopelagic, and bathypelagic communities. The amended epipelagic water mesocosms showed particularly high α- and β-glucosidase activities, which accounted for 4 % to 47 % of the total summed hydrolysis rates, and relatively even peptidase activities (Fig. 1). In contrast, α- and β-glucosidase activities accounted for, at most, ∼ 12 % of the total summed hydrolysis rates in mesopelagic mesocosms and only 3.3 % of the total summed hydrolysis rates in bathypelagic mesocosms. Mesopelagic and bathypelagic mesocosms were typically dominated by high leucine aminopeptidase activities, although this was most notable in bathypelagic mesocosms (Fig. 1).
3.3 Polysaccharide hydrolase activities
Only a limited range of polysaccharides were hydrolyzed in unamended mesocosms. Laminarin and pullulan were hydrolyzed at all depths and stations. However, fucoidan and arabinogalactan, more complex polysaccharides (see “Materials and methods” for additional information), were not hydrolyzed at any depth or station in unamended mesocosms (Figs. 2; S4). Chondroitin was also hydrolyzed in stn. 16 mesopelagic and bathypelagic mesocosms, and xylan was hydrolyzed in stn. 16 epipelagic and mesopelagic mesocosms of both stations (Figs. 2; S4). The spectrum of polysaccharide hydrolase activities was therefore broadest in the mesopelagic. The time course over which individual polysaccharides were hydrolyzed also varied by substrate and depth. In unamended epipelagic and mesopelagic mesocosms, laminarinase activities were generally high at the 2 d time point; pullulanase activities were detected slightly later, except in stn. 16 mesopelagic mesocosms, where pullulanase activity was also measurable at 2 d. Xylanase activity was measurable at 2 d (stn. 12) or at 5 d (stn. 16) in mesopelagic mesocosms; chondroitin hydrolysis was measurable starting at 5 d at stn. 16. The unamended bathypelagic mesocosms showed slightly different patterns, with polysaccharide hydrolase activity detectable starting at 2 d at stn. 16 but only detectable at 17 d at stn. 12 (Fig. S4).

Figure 2Polysaccharide hydrolase activities in amended and unamended mesocosms, averaged between stations. The spectrum of substrates (the range of substrates hydrolyzed) was considerably broader in amended compared to unamended mesocosms. Note that the 0 d time point, when fluorescently labeled polysaccharide incubations began, was 15 d after the addition of HMW organic matter to amended mesocosms. Note the large difference in scales between epi- and mesopelagic mesocosms (up to 40 nmol L−1 h−1) and bathypelagic mesocosms (up to 10 nmol L−1 h−1). Pul: pullulan, Lam: laminarin, Xyl: xylan, Fuc: fucoidan, Ara: arabinogalactan, Chn: chondroitin.
Polysaccharide hydrolase activities were significantly different between amended and unamended mesocosms (ANOVA, p<0.0001). Addition of HMW organic matter increased the rates, broadened the spectrum, and changed the time point at which polysaccharide hydrolase activities were first detected (Fig. 2). All six polysaccharide hydrolase activities were measurable in epipelagic and mesopelagic mesocosms, with the initial time point of detection typically at 2 or 5 d. In amended bathypelagic mesocosms, pullulan, laminarin, and chondroitin hydrolysis were typically initially measured at the 2 or 5 d time points; xylanase activity was usually only detected at the later (17 or 30 d) time points. Maximum polysaccharide hydrolase activities were significantly lower in amended bathypelagic mesocosms (summed activities lower than 20 nmol L−1 h−1) than in epipelagic and mesopelagic mesocosms (summed activities at or above 50 nmol L−1 h−1) (ANOVA, p<0.0001) (Fig. S5). In amended mesocosms, stations did not differ significantly in either the maximum rates (ANOVA, p=0.8401) or the temporal development of polysaccharide hydrolase activities (ANOVA, p=0.7502). Polysaccharide hydrolase activities thus mirror the activities of peptidases and glucosidases, with depth-related differences in enzymatic activities prevailing over location-related differences.
3.4 Bacterial protein production
In amended epipelagic mesocosms, bacterial protein production was high immediately after addition of HMW organic matter (ca. 150 pM h−1) and decreased at subsequent time points (Fig. S6a). In unamended epipelagic mesocosms, bacterial protein production was initially low but increased from 20.6 pM h−1 in stn. 12 mesocosms and 99.3 pM h−1 in stn. 16 mesocosms to rates above 150 pM h−1 at subsequent time points. In mesopelagic mesocosms, bacterial protein production increased in both the amended and unamended mesocosms from very low initial levels to much higher levels at later time points. In stn. 16 mesocosms, bacterial protein production increased with time in both the amended and unamended mesocosms; in stn. 12 mesocosms, the patterns were less clear (Fig. S6a). Bathypelagic amended mesocosms showed detectable bacterial protein production at earlier time points than unamended mesocosms, with rates above 50 pM h−1 at 7 d. Bacterial protein production varied significantly between mesocosms amended with HMW organic matter and unamended mesocosms (ANOVA, p=0.0054), with changes in protein production displaying distinct trends with depth (ANOVA, p<0.0001).
3.5 Bacterial community composition
Initial bacterial communities in epipelagic unamended and amended mesocosms were dominated by Alphaproteobacteria, Cyanobacteria, Flavobacteriia, and Gammaproteobacteria, as demonstrated by relative read abundance (Figs. 3; S7). In meso- and bathypelagic mesocosms, Alphaproteobacteria, Deltaproteobacteria, Gammaproteobacteria, Flavobacteriia, and the SAR202 clade dominated initial bacterial communities, although these classes were present in different relative proportions in mesopelagic and bathypelagic mesocosms (Figs. 3; S7). As expected, initial (t0) bacterial communities in amended and unamended mesocosms were not significantly different from one another (PERMANOVA, R2=0.02016, p=0.655). Distinct bacterial communities were present in mesocosms from different depths (Figs. 4; S8), and depth explained the greatest amount of dissimilarity between bacterial communities (PERMANOVA, R2=0.1968, p=0.001). Samples from comparable depths clustered together, regardless of their station of origin (shown by non-metric multidimensional scaling (NMDS) based on the Bray–Curtis dissimilarity index; Figs. 4; S8). However, samples collected from the same water masses but at different stations still displayed differences in community composition that were statistically significant (PERMANOVA, R2=0.0190, p=0.001); we have therefore graphed community composition at each station separately (Figs. 3; S7). Duplicate filters, both sample duplicates and filter duplicates from the same depth and station, displayed consistent reproducibility of bacterial community composition: hierarchical clustering showed that duplicates clustered closest together, except in a single case (Fig. S9).
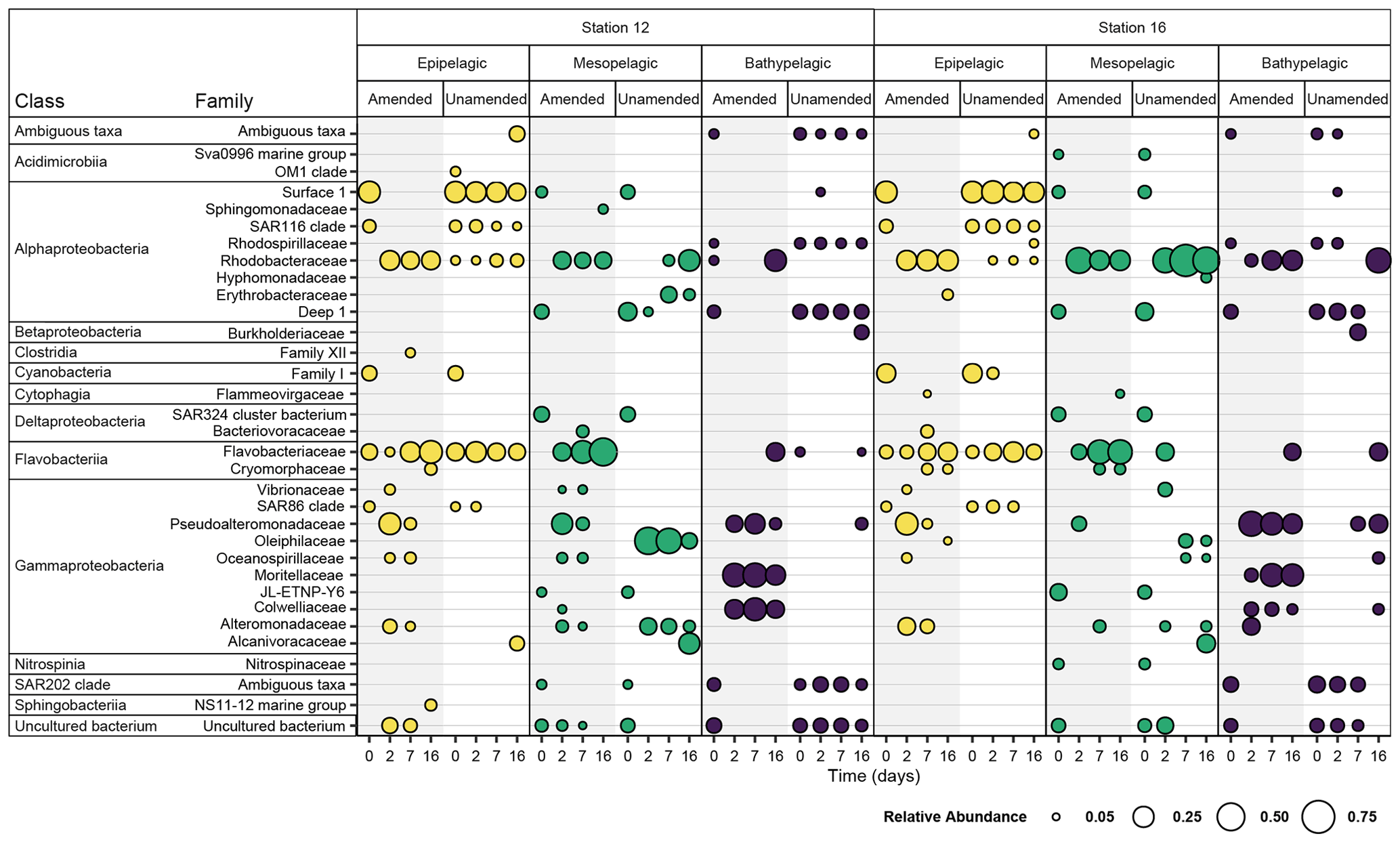
Figure 3Bacterial families with a normalized relative abundance of 5 % or more in amended and unamended mesocosms from each station and depth 0, 2, 7, and 16 d after the addition of HMW organic matter to amended mesocosms. Amended values are an average of triplicate amended mesocosms, and the bubble size indicates the relative abundance (%) of each bacterial family. Note that “Ambiguous taxa” represents those sequences for which the taxonomy did not meet a consensus when compared to the reference database.
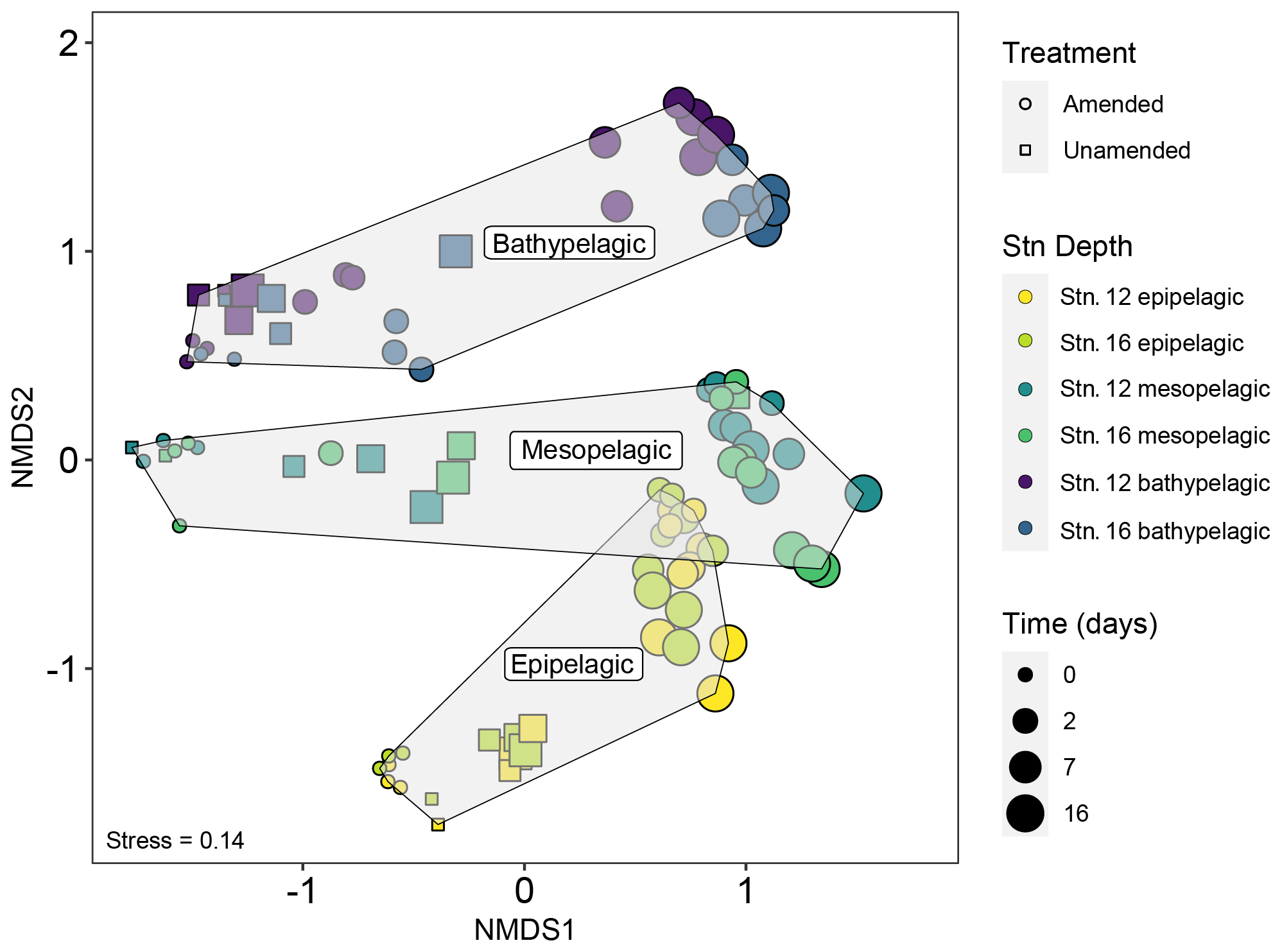
Figure 4Non-metric multidimensional scaling (NMDS) plot of bacterial community composition based on the Bray–Curtis dissimilarity index shows that communities were quite distinct by depth. In later phases of the incubations, however, the amended mesopelagic and epipelagic mesocosms become more similar to one another. Note also that mesocosms from stns. 12 and 16 from a given depth and time point – especially for epipelagic and mesopelagic samples – remained similar to one another through the course of the incubations.
Addition of HMW organic matter resulted in distinct shifts in bacterial community composition, which were evident starting at 2 d (Figs. 3, 4, S7). While initial bacterial communities from the mesopelagic were more compositionally and phylogenetically similar to bathypelagic communities (Figs. S7, S10), after HMW organic matter addition, mesopelagic communities progressively became more similar to bacterial communities in epipelagic mesocosms. This shift was the result of similar increases in the relative proportions of Gammaproteobacteria and Flavobacteriia, as well as a decrease in the relative proportion of Alphaproteobacteria, in amended epipelagic and mesopelagic mesocosms (Figs. 3, S7). Relative read abundances for Flavobacteriia and Alphaproteobacteria in bathypelagic mesocosms were notably lower than in epipelagic and mesopelagic mesocosms, while Gammaproteobacteria relative read abundance was higher. The composition of bacterial communities in unamended mesocosms also shifted with time, showing evidence of a “bottle effect”. However, these compositional shifts were observed to a lesser extent (Figs. 3, S7) and, especially for bathypelagic mesocosms, occurred at later time points than in their amended counterparts (Figs. 3, S7).
Within the Gammaproteobacteria, families that became abundant after addition of HMW organic matter were initially present in very low relative proportions and, in some cases, were not detected during initial time points (Fig. S11). The gammaproteobacterial families with the highest read abundance varied with depth, with taxa in the Alteromonadaceae, Colwelliaceae, Oceanospirillaceae, Pseudoalteromonadaceae, and Vibrionaceae families becoming abundant in epipelagic and mesopelagic mesocosms, while bathypelagic mesocosms were dominated by taxa in the Alteromonadaceae, Colwelliaceae, Moritellaceae, and Pseudoalteromonadaceae families (Fig. S11). Although members of the Alteromonadaceae, Colwelliaceae, and Pseudoalteromonadaceae were present in amended mesocosms from all depths, their relative proportion varied with depth: the highest relative proportion of Alteromonadaceae typically occurred in epipelagic and mesopelagic mesocosms, while Colwelliaceae were most abundant in bathypelagic mesocosms; the relative proportion of Pseudoalteromonadaceae did not follow a consistent depth-related pattern (Fig. S11). In some cases, a single genus or OTU within these families became abundant (Figs. S12, S13); for example, in the Colwelliaceae family, an OTU in the genus Colwellia became abundant in all amended mesocosms (Fig. S14).
The presence of these gammaproteobacterial families in amended mesocosms contrasts sharply with the dominant families in unamended mesocosms: in epipelagic water unamended mesocosms, the SAR86 clade remained the dominant taxa throughout most time points, while the proportion of Pseudoalteromonadaceae and Oceanospirillaceae remained low. In unamended bathypelagic mesocosms, taxa that were initially highly abundant, such as the Salinisphaeraceae, also typically comprised a large proportion of all taxa during subsequent time points (Fig. S11). However, in unamended mesopelagic mesocosms, relative gammaproteobacterial read abundance shifted considerably after the initial time point. The taxa that became abundant were present, albeit at low relative abundances, during initial time points, and the patterns in the abundance of these taxa differed between stations.
While distinct families in the Gammaproteobacteria became abundant in water from different depths, the same families in the Alphaproteobacteria and Flavobacteriia became dominant in all amended mesocosms after t0, regardless of depth. Within the Flavobacteriia, members of the Flavobacteriaceae family increased in relative read abundance in all mesocosms after t0, although they were generally less abundant in bathypelagic mesocosms than in epipelagic and mesopelagic mesocosms (Fig. S15). The relative proportion of other Flavobacteriia families, including the Cryomorphaceae and the NS9 marine group, decreased over time. The same trend was observed in all unamended mesocosms (Fig. S15). Although the Flavobacteriaceae family became abundant in all amended mesocosms, the dominant Flavobacteriaceae genera varied greatly with depth (Fig. S16). For example, Polaribacter 4, although present to some degree in all amended mesocosms, was the most abundant Flavobacteriaceae genera in bathypelagic mesocosms, while Tenacibaculum was more abundant in epipelagic and mesopelagic mesocosms (Fig. S16). In the Alphaproteobacteria, the Rhodobacteraceae became dominant in all amended mesocosms, despite only making up a low relative proportion of Alphaproteobacteria families initially (Fig. S16). A shift in the relative proportions of alphaproteobacterial families occurred after 2 d in epipelagic and mesopelagic mesocosms, as well as after 7 d in bathypelagic mesocosms, when the Rhodobacteraceae became the dominant alphaproteobacterial family (Fig. S17). Within the Rhodobacteraceae family, bacteria in the genus Sulfitobacter became particularly abundant in all amended mesocosms (Fig. S18).
Bacterial richness, evenness, and Shannon diversity typically decreased in all amended mesocosms 2 d after the addition of HMW organic matter; this decrease was greatest in water from the meso- and bathypelagic zones (Fig. S6). However, in epipelagic and mesopelagic mesocosms, these decreases in diversity were not significantly different from those in unamended mesocosms (ANOVA, p>0.05 for all, Table S2). Only in bathypelagic mesocosms were changes in richness, evenness, and diversity significantly different between amended and unamended mesocosms (ANOVA, p<0.005, Table S2): while richness, evenness, and diversity in amended bathypelagic mesocosms had decreased significantly 2 d after the addition of HMW organic matter, the decrease in these parameters in unamended mesocosms was more gradual and in some cases even increased or remained relatively consistent (Fig. S6).
3.6 Relating community composition to enzymatic activities
Successional patterns in bacterial community composition were concurrent with functional changes in enzymatic capabilities. Notable changes in peptidase and glucosidase activities in epipelagic and mesopelagic mesocosms at the 2 d time point corresponded with a major shift in bacterial community composition (Figs. 1, 4), with Gammaproteobacteria and Flavobacteriia becoming highly abundant (Fig. 3). In bathypelagic mesocosms, a shift of similar magnitude was measured 7 d after the addition of HMW organic matter (Fig. 4). Since the unamended mesocosms did not show comparable shifts in enzyme activities or community composition, a major driver of the shifts in amended mesocosms was most likely the addition of HMW organic matter, rather than bottle effects. The relationship between bacterial community composition and peptidase and glucosidase activity in amended mesocosms after 2 d was stronger and of greater significance (Mantel R=0.1483, p=0.003) than in unamended mesocosms and amended mesocosms at t0, when the relationship between bacterial community composition and peptidase and glucosidase activity was not significant (Mantel R=0.0080, p=0.356).
Distinct depth-related differences in enzyme function and community composition characterized the epipelagic, mesopelagic, and bathypelagic mesocosms: with increasing depth, the rates and/or spectrum of substrates hydrolyzed decreased, consistent with previous measurements of enzymatic activities in unamended water (Hoarfrost and Arnosti, 2017; Balmonte et al., 2018; Hoarfrost et al., 2019). Addition of HMW organic matter, however, revealed depth-related responses that were not evident in the unamended mesocosms: not only the rates but also the spectrum of enzyme activities broadened in amended mesocosms, but the extent to which this broader spectrum was measurable varied by depth, with the amended bathypelagic microbial communities in particular showing a less-broad response than their mesopelagic and epipelagic counterparts (Fig. 5).
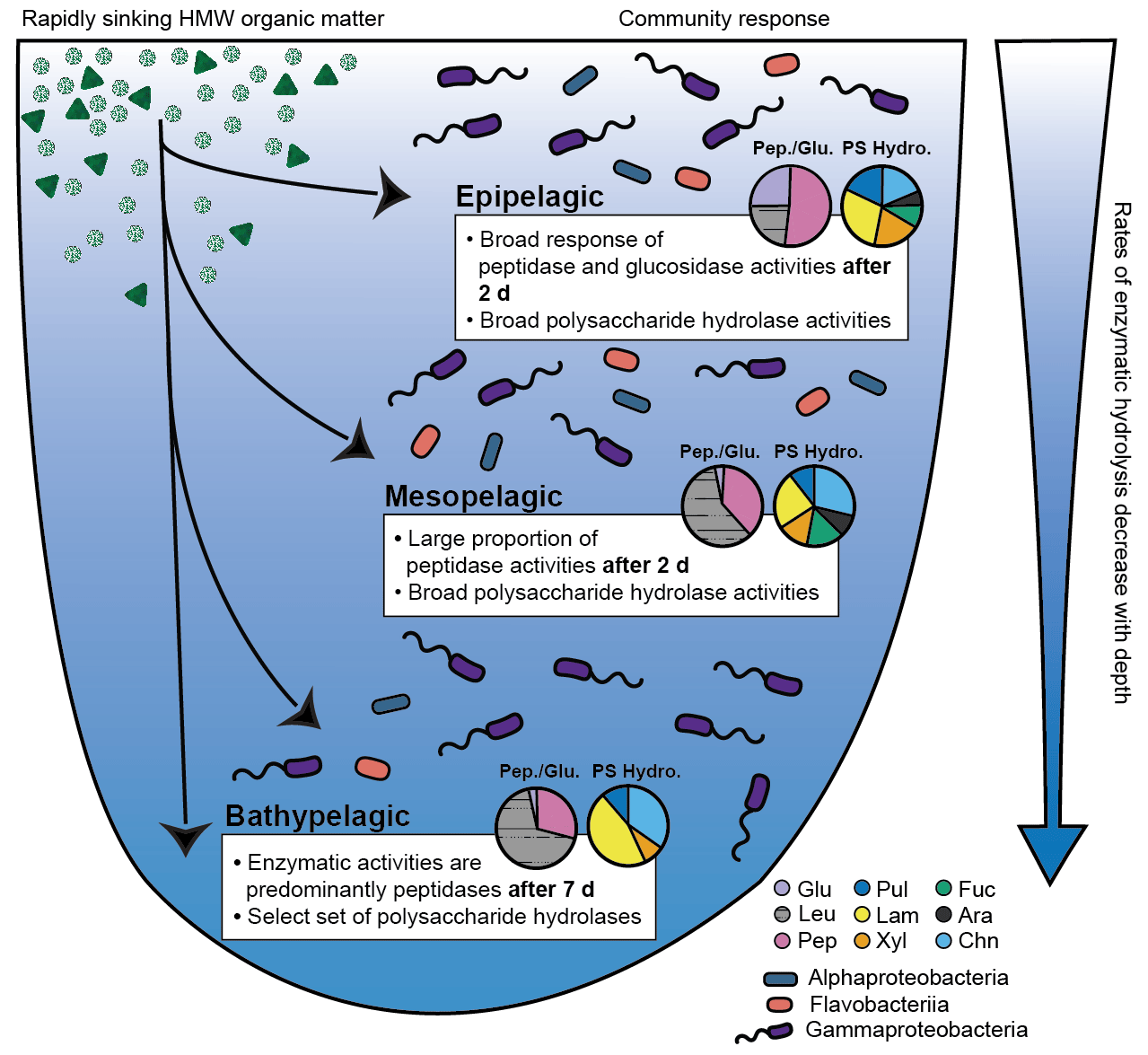
Figure 5Conceptual figure illustrates the response of bacterial communities in North Atlantic epipelagic, mesopelagic, and bathypelagic waters to an input of rapidly sinking, fresh, HMW organic matter. Community responses depicted here are the time point at which significant responses to the input of HMW organic matter was measured (2 d in epipelagic and mesopelagic incubations and 7 d in bathypelagic incubations); polysaccharide hydrolase activities are an average of all time points. Pie charts show the relative contributions of peptidases and glucosidases (labeled as Pep./Glu.) and polysaccharide hydrolases (labeled as PS Hydro.). Glu: α- and β-glucosidases, Leu: leucine aminopeptidases, Pep: trypsin and chymotrypsin activities, Pul: pullulanases, Lam: laminarinases, Xyl: xylanases, Fuc: fucoidanases, Ara: arabinogalactanases, Chn: chondroitin sulfate hydrolases. The relative abundance of bacterial communities is illustrated by the three major classes of bacteria that responded to HMW organic matter input (Alphaproteobacteria, Flavobacteriia, and Gammaproteobacteria).
These depth-related functional differences were similar between comparable depths at two stations separated by 1370 km (Figs. S1, S3, S4). Furthermore, the patterns were also very similar to those measured in epipelagic and bathypelagic mesocosms amended with the same HMW organic matter at a different open-ocean station in the western North Atlantic Ocean during the previous year (Balmonte et al., 2019; stn. 8, water depth 4574 m; Fig. S1). These results suggest that the response of bacterial communities to an input of HMW organic matter – both in terms of community composition and enzymatic function – varies far more by depth than location and may be predictable over a regional scale within individual water masses at similar depths.
These robust enzymatic patterns may also indicate the “value” of specific substrates to a microbial community in each depth zone. Amendment of epipelagic waters, for example, led to very high α- and β-glucosidase activities, which were higher by several orders of magnitude than rates typically measured in unamended epipelagic waters (Baltar et al., 2009, 2010; Hoarfrost and Arnosti, 2017; Hoarfrost et al., 2019). This pattern suggests that removal of terminal glucose units, whether from polysaccharides, glycosylated proteins, or lipids, may be a critical first step in accessing the complex structures “underneath” (Fig. 5). This feature was also evident during the previous year in epipelagic mesocosms containing water from stn. 8 (Fig. S1; Balmonte et al., 2019). Initial removal of terminal glucose and leucine from a HMW substrate may in effect clear the way for activities of a broad range of endopeptidases and polysaccharide hydrolases, which cleave structures mid-chain. The rapid response of the epipelagic community across the entire range of enzyme activities, as well as the high hydrolysis rates triggered by substrate addition, may reflect the fact that microbial communities in epipelagic waters are frequently exposed to freshly produced phytoplankton-derived organic matter rich in amino acids and carbohydrates (Benner et al., 1997; Wakeham et al., 1997; Kaiser and Benner, 2012).
The bacterial taxa in epipelagic mesocosms that responded to the addition of HMW organic matter by the 2 d time point, in particular increases in the abundance of Rhodobacteraceae (Alphaproteobacteria) and Pseudoalteromonadaceae (Gammaproteobacteria) (Fig. 3), also reflect changes in community composition seen in the ocean: bacterial taxa within these classes are usually rare or present in low abundances in seawater but respond rapidly to seasonal changes in organic matter abundance, such as during phytoplankton blooms (Teeling et al., 2012; Fuhrman et al., 2015; Avcı et al., 2020). We hypothesize that the Rhodobacteraceae in particular were likely responding to an increase in low-molecular-weight substrates that could have been released through the activities of the glucosidases and peptidases, enzymes potentially produced by Pseudoalteromonadaceae (Alonso-Sáez et al., 2012; Li et al., 2018).
In mesopelagic mesocosms, the addition of HMW organic matter also produced a broad enzymatic response, consistent with observations of considerable organic matter transformation in this zone (Wakeham et al., 1997; Benner and Amon, 2015). However, mesopelagic mesocosms showed a less robust glucosidase response than epipelagic mesocosms and were dominated by leucine aminopeptidase, an exopeptidase activity that may integrate the activities of a range of exopeptidases (Steen et al., 2015) (Fig. 5). Enhancement of rates of peptidase and glucosidase activities were considerably lower than in the epipelagic zone (Fig. 1). The similarities in polysaccharide hydrolase activities in the epipelagic and mesopelagic mesocosms (Fig. 2) may be due in part to convergent trends in bacterial community composition over time (Figs. 4, S8, S10), since the polysaccharide hydrolase measurements tested the enzymatic responses of microbial communities after they had considerable time to react to substrate addition (Fig. 5). Together, this evidence suggests that mesopelagic communities can respond rapidly to inputs of fresh HMW organic matter, though the rate and spectrum of enzymatic activities differs from those in the epipelagic zone (Fig. 5).
The bathypelagic microbial communities responded in a markedly more limited manner. The spectrum of enzyme activities was less broad, and the increases in the rate of enzymatic activities were less substantial and occurred over longer timescales than in epipelagic and mesopelagic communities (Figs. 1, 2). The differences in response times between the amended epipelagic and mesopelagic communities relative to bathypelagic communities may be related in part to differences in the initial size and activity of the heterotrophic bacterial populations. For example, bathypelagic bacterial communities had significantly lower rates of protein production than epipelagic and mesopelagic communities, and protein production rates in amended bathypelagic mesocosms did not increase as quickly as those of mesopelagic and epipelagic communities (Fig. S6a). This slower response may suggest that members of these bathypelagic communities were dormant due to limited carbon availability. Fluxes of organic matter that reach the bathypelagic zone may be sporadic in nature, so bathypelagic communities can enter dormancy until fresh organic matter becomes available, responding even after long periods of starvation (Sebastián et al., 2019). These communities would thus require additional time (relative to epipelagic and mesopelagic communities) to respond to pulses of organic matter, resulting in slower enzymatic responses. Differences in temperature between the incubations (Table 1) may have also played a role in differences in enzymatic activities between the three depths; however, the observation that some enzyme activities were detectable only in amended mesocosms and others were not detectable at all over the entire time course of the experiments (Figs. 1, 2) suggests that the absence of some enzyme activities in bathypelagic samples is not simply a function of temperature.
In bathypelagic mesocosms, increases in bacterial protein production (Fig. S6a) and the rates of peptidase and glucosidase activities (Fig. 1) at the 7 d time point also corresponded with a shift in bacterial community composition equivalent to that in epipelagic and mesopelagic mesocosms after 2 d (Fig. 4). The substantial decrease in bacterial diversity, richness, and evenness that coincided with this shift in bacterial community composition was likely a result of selection for bacteria capable of degrading HMW organic matter. This shift in community composition resulted in increases in the abundance of Moritellaceae (Gammaproteobacteria), Colwelliaceae (Gammaproteobacteria), Pseudoalteromonadaceae (Gammaproteobacteria), and Rhodobacteraceae (Alphaproteobacteria) families that had previously been observed responding to the same HMW organic matter in bathypelagic mesocosms (Balmonte et al., 2019). Although these families were present in epipelagic and mesopelagic mesocosms, their response and abundance differed greatly from that in bathypelagic mesocosms, where they became the dominant taxa after the addition of HMW organic matter (Fig. 3). This variability of community composition with depth may account for some of the strong functional differences we observed between distinct bacterial communities, as the ability to produce the enzymes necessary to degrade HMW organic matter varies widely among different bacterial taxa, even those that are closely related (Xing et al., 2015; Saw et al., 2020; Avcı et al., 2020).
The narrower spectrum of polysaccharide hydrolase activities measured in amended bathypelagic mesocosms suggests that the enzymatic investment to hydrolyze some of the complex structures that were degraded at shallower depths is not likely to pay off for bathypelagic microbial communities. At stns. 12 and 16, neither fucoidan or arabinogalactan were hydrolyzed, consistent with results from bathypelagic enzyme activities in amended mesocosms from stn. 8 (Fig. 2; Balmonte et al., 2019). This limited spectrum of extracellular enzymatic activities may reflect their energetic cost–benefit balance – as both arabinogalactan and fucoidan are structurally complex and likely more recalcitrant to microbial degradation (Sichert et al., 2020; Vidal-Melgosa et al., 2021), the cost of enzymatic expression may be too high to justify “investment” in the tools needed to hydrolyze certain structures. Hydrolysis of fucoidan, for example, requires high levels of specialization and energetic investment into hundreds of enzymes (Sichert et al., 2020). If the frequency of encounter with fucoidan in the bathypelagic zone is low, this strategy may not be productive, as the production of extracellular enzymes is profitable only when the return on investment is sufficient (e.g., Traving et al., 2015). Therefore, the lack of measurable fucoidanase and arabinogalactanase activities in bathypelagic waters suggests that these substrates may not be available in sufficiently high concentrations for production of the required enzymes to pay off. However, the four polysaccharides (pullulan, laminarin, xylan, and chondroitin) that were hydrolyzed in amended bottom water mesocosms represent a breadth and rate of polysaccharide hydrolase activities not typically measurable in bulk incubations of bathypelagic water (Fig. 5; Hoarfrost and Arnosti, 2017; Balmonte et al., 2018, 2021). In any case, the enhanced capability of amended bathypelagic microbial communities to degrade complex polysaccharides suggests that an influx of fresh organic matter to the bathypelagic zone (Broek et al., 2020; Poff et al., 2021) may fuel the growth of a select set of organisms enzymatically equipped to take advantage of this resource (Fig. 5).
Our measurements of depth-related differences in enzymatic activities between distinct microbial communities from the epipelagic, mesopelagic, and bathypelagic zones in response to an addition of the same HMW organic matter were consistent over a regional scale. The diverse enzymatic responses of epi-, meso-, and bathypelagic microbial communities to inputs of HMW organic matter imply that the structure of HMW organic matter, its residence time at specific depths in the water column, and the distinct enzymatic capabilities of heterotrophic microbial communities at different depths are key factors controlling the ultimate fate of organic matter in the ocean.
Raw data for peptidase, glucosidase, and polysaccharide hydrolase activities are available through BCO-DMO (https://www.bco-dmo.org/project/712359, last access: 20 October 2017; https://doi.org/10.26008/1912/bco-dmo.717495.1, Arnosti, 2018), and 16S rRNA sequence data can be accessed at the NCBI Sequence Read Archive under the accession number PRJNA480640.
The supplement related to this article is available online at: https://doi.org/10.5194/bg-19-5617-2022-supplement.
CA designed the experiments and coordinated work at sea. SAB, AH, JPB, and SG collected and processed samples at sea and ashore. SAB extracted DNA, processed and analyzed the sequence data, and conducted statistical analyses. SAB and CA analyzed results, wrote the manuscript, and revised it with input from all co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank the captain, crew, and scientific party of R/V Endeavor, cruise EN584. Karylle Abella assisted in sample collection and processing, John Bane helped to interpret the physiochemical data, and Jakub Kwintkiewicz aided in library preparation and sequencing of the 16S rRNA samples. We would also like to thank James Umbanhowar for his advice on statistical analyses.
This research has been supported by the National Science Foundation (grant nos. OCE-1332881 and OCE-2022952).
This paper was edited by Andrew Thurber and reviewed by two anonymous referees.
Alderkamp, A. C., Van Rijssel, M., and Bolhuis, H.: Characterization of marine bacteria and the activity of their enzyme systems involved in degradation of the algal storage glucan laminarin, FEMS Microbiol. Ecol., 59, 108–117, https://doi.org/10.1111/j.1574-6941.2006.00219.x, 2007.
Alonso-Sáez, L., Sánchez, O., and Gasol, J. M.: Bacterial uptake of low molecular weight organics in the subtropical Atlantic: Are major phylogenetic groups functionally different?, Limnol. Oceanogr., 57, 798–808, https://doi.org/10.4319/lo.2012.57.3.0798, 2012.
Amon, R. M., Fitznar, H. P., and Benner, R.: Linkages among the bioreactivity, chemical composition, and diagenetic state of marine dissolved organic matter, Limnol. Oceanogr., 46, 287–297, https://doi.org/10.4319/lo.2001.46.2.0287, 2001.
Araki, T., Hashikawa, S., and Morishita, T.: Cloning, sequencing, and expression in Escherichia coli of the new gene encoding b-1,3-xylanase from a marine bacterium, Vibrio sp. Strain XY-214, Appl. Environ. Microb., 66, 1741–1743, 2000.
Arnosti, C.: Fluorescent derivatization of polysaccharides and carbohydrate-containing biopolymers for measurement of enzyme activities in complex media, J. Chromatogr. B., 793, 181–191, https://doi.org/10.1016/S1570-0232(03)00375-1, 2003.
Arnosti, C.: Microbial extracellular enzymes and the marine carbon cycle, Annu. Rev. Mar. Sci., 3, 401–425, https://doi.org/10.1146/annurev-marine-120709-142731, 2011.
Arnosti, C.: Project: Latitudinal and depth-related contrasts in enzymatic capabilities of pelagic microbial communities: Predictable patterns in the ocean?, BCO-DMO [data set], https://doi.org/10.26008/1912/bco-dmo.717495.1, 2018.
Arnosti, C. and Repeta, D. J.: Extracellular enzyme activity in anaerobic bacterial cultures: Evidence of pullulanase activity among mesophilic marine bacteria, Appl. Environ. Microb., 60, 840–846, 1994.
Arnosti, C., Steen, A. D., Ziervogel, K., Ghobrial, S., and Jeffrey, W. H.: Latitudinal gradients in degradation of marine dissolved organic carbon, PLoS One, 6, e28900, https://doi.org/10.1371/journal.pone.0028900, 2011.
Avcı, B., Krüger, K., Fuchs, B. M., Teeling, H., and Amann, R. I.: Polysaccharide niche partitioning of distinct Polaribacter clades during North Sea spring algal blooms, ISME J., 14, 1–15, https://doi.org/10.1038/s41396-020-0601-y, 2020.
Azam, F.: Microbial control of oceanic carbon flux: the plot thickens, Science, 280, 694–696, https://doi.org/10.1126/science.280.5364.694, 1998.
Balmonte, J. P., Teske, A., and Arnosti, C.: Structure and function of high Arctic pelagic, particle-associated and benthic bacterial communities, Environ. Microbiol., 20, 2941–2954, https://doi.org/10.1111/1462-2920.14304, 2018.
Balmonte, J. P., Buckley, A., Hoarfrost, A., Ghobrial, S., Ziervogel, K., Teske, A., and Arnosti, C.: Community structural differences shape microbial responses to high molecular weight organic matter, Environ. Microbiol., 21, 557–571, https://doi.org/10.1111/1462-2920.14485, 2019.
Balmonte, J. P., Simon, M., Giebel, H. A., and Arnosti, C.: A sea change in microbial enzymes: Heterogeneous latitudinal and depth-related gradients in bulk water and particle-associated enzymatic activities from 30∘ S to 59∘ N in the Pacific Ocean, Limnol. Oceanogr., 66, 3489–3507, https://doi.org/10.1002/lno.11894, 2021.
Baltar, F., Arístegui, J., Sintes, E., Van Aken, H. M., Gasol, J. M., and Herndl, G. J.: Prokaryotic extracellular enzymatic activity in relation to biomass production and respiration in the meso-and bathypelagic waters of the (sub) tropical Atlantic, Environ. Microbiol., 11, 1998–2014, https://doi.org/10.1111/j.1462-2920.2009.01922.x, 2009.
Baltar, F., Arístegui, J., Gasol, J. M., Sintes, E., van Aken, H. M., and Herndl, G. J.: High dissolved extracellular enzymatic activity in the deep central Atlantic Ocean, Aquat. Microb. Ecol., 58, 287–302, https://doi.org/10.3354/ame01377, 2010.
Becker, S., Tebben, J., Coffinet, S., Wiltshire, K., Iversen, M. H., Harder, T., Hinrichs, K. U., and Hehemann, J. H.: Laminarin is a major molecule in the marine carbon cycle, P. Natl. Acad. Sci. USA, 117, 6599–6607, https://doi.org/10.1073/pnas.1917001117, 2020.
Beier, S., Rivers, A. R., Moran, M. A., and Obernosterer, I.: The transcriptional response of prokaryotes to phytoplankton-derived dissolved organic matter in seawater, Environ. Microbiol., 17, 3466–3480, https://doi.org/10.1111/1462-2920.12434, 2015.
Benner, R. and Amon, R. M.: The size-reactivity continuum of major bioelements in the ocean, Annu. Rev. Mar. Sci., 7, 185–205, https://doi.org/10.1146/annurev-marine-010213-135126, 2015.
Benner, R., Biddanda, B., Black, B., and McCarthy, M.: Abundance, size distribution, and stable carbon and nitrogen isotopic compositions of marine organic matter isolated by tangential-flow ultrafiltration, Mar. Chem., 57, 243–263, https://doi.org/10.1016/S0304-4203(97)00013-3, 1997.
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C., Al-Ghalith, G. A., Alexander, H., Alm, E. J., Arumugam, M., Asnicar, F., and Bai, Y.: Reproducible, interactive, scalable and extensible microbiome data using QIIME 2, Nat. Biotechnol., 37, 852–857, https://doi.org/10.1038/s41587-019-0209-9, 2019.
Broek, T. A., Walker, B. D., Guilderson, T. P., Vaughn, J. S., Mason, H. E., and McCarthy, M. D.: Low molecular weight dissolved organic carbon: aging, compositional changes, and selective utilization during global ocean circulation, Global Biogeochem. Cy., 34, e2020GB006547, https://doi.org/10.1029/2020GB006547, 2020.
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P.: DADA2: high-resolution sample inference from Illumina amplicon data, Nat. Methods, 13, 581–583, https://doi.org/10.1038/nmeth.3869, 2016.
DeLong, E. F., Preston, C. M., Mincer, T., Rich, V., Hallam, S. J., Frigaard, N. U., Martinez, A., Sullivan, M. B., Edwards, R., Brito, B. R., and Chisholm, S. W.: Community genomics among stratified microbial assemblages in the ocean's interior, Science, 311, 496–503, https://doi.org/10.1126/science.1120250, 2006.
Fuhrman, J. A., Cram, J. A., and Needham, D. M.: Marine microbial community dynamics and their ecological interpretation, Nat. Rev. Microbiol., 13, 133–146, https://doi.org/10.1038/nmeth.3869, 2015.
Guerrero-Feijóo, E., Nieto-Cid, M., Sintes, E., Dobal-Amador, V., Hernando-Morales, V., Álvarez, M., Balagué, V., and Varela, M. M.: Optical properties of dissolved organic matter relate to different depth-specific patterns of archaeal and bacterial community structure in the North Atlantic Ocean, FEMS Microbiol. Ecol., 93, fiw224, https://doi.org/10.1093/femsec/fiw224, 2016.
Hartley, B., Barber, H. G., Carter, J. R., and Sims, P. A.: An atlas of British diatoms, Balogh Scientific Books, ISBN 9780948737459, 1996.
Hedges, J. I., Baldock, J. A., Gelinas, Y., Lee, C., Peterson, M., and Wakeham, S. G.: Evidence for non-selective preservation of organic matter in sinking particlesm, Nature 409, 801–804, 2001.
Heiderich, J. and Todd, R. E.: Along-stream evolution of Gulf Stream volume transport, J. Phys. Oceanogr., 50, 2251–2270, https://doi.org/10.1175/JPO-D-19-0303.1, 2020.
Hoarfrost, A. and Arnosti, C.: Heterotrophic Extracellular Enzymatic Activities in the Atlantic Ocean Follow Patterns Across Spatial and Depth Regimes, Front. Mar. Sci., 4, 200, https://doi.org/10.3389/fmars.2017.00200, 2017.
Hoarfrost, A., Balmonte, J. P., Ghobrial, S., Ziervogel, K., Bane, J., Gawarkiewicz, G., and Arnosti, C.: Gulf Stream ring water intrusion on the Mid-Atlantic Bight continental shelf break affects microbially-driven carbon cycling, Front. Mar. Sci., 6, 394, https://doi.org/10.3389/fmars.2019.00394, 2019.
Hoppe, H. G.: Significance of exoenzymatic activities in the ecology of brackish water: measurements by means of methylumbelliferyl-substrates, Marine Ecol.-Prog. Ser., 11, 299–308, https://www.jstor.org/stable/44634717 (last access: 28 November 2022), 1983.
Kaiser, K. and Benner, R.: Organic matter transformations in the upper mesopelagic zone of the North Pacific: Chemical composition and linkages to microbial community structure, J. Geophys. Res.-Oceans, 117, C01023, https://doi.org/10.1029/2011JC007141, 2012.
Kirchman, D.: Measuring bacterial biomass production and growth rates from leucine incorporation in natural aquatic environments, Method Microbiol., 30, 227–237, https://doi.org/10.1016/S0580-9517(01)30047-8, 2001.
Kirchman, D., K'Nees, E., and Hodson, R.: Leucine incorporation and its potential as a measure of protein synthesis by bacteria in natural aquatic systems, Appl. Environ. Microb., 49, 599–607, https://doi.org/10.1128/aem.49.3.599-607.1985, 1985.
Li, D. X., Zhang, H., Chen, X. H., Xie, Z. X., Zhang, Y., Zhang, S. F., Lin, L., Chen, F., and Wang, D. Z.: Metaproteomics reveals major microbial players and their metabolic activities during the blooming period of a marine dinoflagellate Prorocentrum donghaiense, Environ. Microbiol., 20, 632–644, https://doi.org/10.1111/1462-2920.13986, 2018.
Luria, C. M., Amaral-Zettler, L. A., Ducklow, H. W., Repeta, D. J., Rhyne, A. L., and Rich, J. J.: Seasonal shifts in bacterial community responses to phytoplankton-derived dissolved organic matter in the Western Antarctic Peninsula, Front. Microbiol., 8, 2117, https://doi.org/10.3389/fmicb.2017.02117, 2017.
Martin, M.: Cutadapt removes adapter sequences from high-throughput sequencing reads, EMBnet J., 17, 1–10, 2011.
McMurdie, P. J. and Holmes, S.: phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data, PloS one, 8, e61217, https://doi.org/10.1371/journal.pone.0061217, 2013.
Mestre, M., Ruiz-González, C., Logares, R., Duarte, C. M., Gasol, J. M., and Sala, M. M.: Sinking particles promote vertical connectivity in the ocean microbiome, P. Natl. Acad. Sci. USA, 115, E6799–E6807, https://doi.org/10.1073/pnas.1802470115, 2018.
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., Minchin, P. R., O'Hara, R. B., Simpson, G. L., Solymos, R., Stevens, M. H. H., Szoecs, E., Wagner, H.: vegan: Community Ecology Package, R package version 2.4-6, https://CRAN.R-project.org/package=vegan, last access: 24 August 2018.
Pinheiro, J., Bates, D., DebRoy, S., Sarkar, D., and R Core Team: nlme: Linear and Nonlinear Mixed Effects Models, R package version 3.1-137, https://CRAN.R-project.org/package=nlme, last access: 24 August 2018.
Poff, K. E., Leu, A. O., Eppley, J. M., Karl, D. M., and DeLong, E. F.: Microbial dynamics of elevated carbon flux in the open ocean's abyss, P. Natl. Acad. Sci.-Biol., 118, e2018269118, https://doi.org/10.1073/pnas.2018269118, 2021.
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., and Glöckner, F. O.: SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB, Nucleic Acids Res., 35, 7188–7196, https://doi.org/10.1093/nar/gkm864, 2007.
R Core Team: R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org/ (last access: 12 October 2022), 2017.
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F.: VSEARCH: a versatile open source tool for metagenomics, PeerJ, 4, e2584, https://doi.org/10.7717/peerj.2584, 2016.
Ruiz-González, C., Mestre, M., Estrada, M., Sebastián, M., Salazar, G., Agustí, S., Moreno-Ostos, E., Reche, I., Álvarez-Salgado, X. A., Morán, X. A., and Duarte, C. M.: Major imprint of surface plankton on deep ocean prokaryotic structure and activity, Mol. Ecol., 29, 1820–1838, https://doi.org/10.1111/mec.15454, 2020.
Saw, J. H., Nunoura, T., Hirai, M., Takaki, Y., Parsons, R., Michelsen, M., Longnecker, K., Kujawinski, E. B., Stepanauskas, R., Landry, Z., and Carlson, C. A.: Pangenomics Analysis Reveals Diversification of Enzyme Families and Niche Specialization in Globally Abundant SAR202 Bacteria, mBio, 11, e02975-19, https://doi.org/10.1128/mBio.02975-19, 2020.
Sebastián, M., Estrany, M., Ruiz-González, C., Forn, I., Sala, M. M., Gasol, J. M., and Marrasé, C.: High growth potential of long-term starved deep ocean opportunistic heterotrophic bacteria, Front. Microbiol., 10, 760, https://doi.org/10.3389/fmicb.2019.00760, 2019.
Sebastián, M., Forn, I., Auladell, A., Gómez-Letona, M., Sala, M. M., Gasol, J. M., and Marrasé, C.: Differential recruitment of opportunistic taxa leads to contrasting abilities in carbon processing by bathypelagic and surface microbial communities, Environ. Microbiol., 23, 190–206, https://doi.org/10.1111/1462-2920.15292, 2021.
Sichert, A., Corzett, C. H., Schechter, M. S., Unfried, F., Markert, S., Becher, D., Fernandez-Guerra, A., Liebeke, M., Schweder, T., Polz, M. F., and Hehemann, J. H.: Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan, Nat. Microbiol., 5, 1026–1039, https://doi.org/10.1038/s41564-020-0720-2, 2020.
Simon, M. and Azam, F.: Protein content and protein synthesis rates of planktonic marine bacteria, Mar. Ecol.-Prog. Ser., 51, 201–213, 1989.
Steen, A. D., Ziervogel, K., Ghobrial, S., and Arnosti, C.: Functional variation among polysaccharide-hydrolyzing microbial communities in the Gulf of Mexico, Mar. Chem., 138, 13–20, https://doi.org/10.1016/j.marchem.2012.06.001, 2012.
Steen, A. D., Vazin, J. P., Hagen, S. M., Mulligan, K. H., and Wilhelm, S. W.: Substrate specificity of aquatic extracellular peptidases assessed by competitive inhibition assays using synthetic substrates, Aquat. Microb. Ecol., 75, 271–281, https://doi.org/10.3354/ame01755, 2015.
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., Djahanschiri, B., Zeller, G., Mende, D. R., Alberti, A., and Cornejo-Castillo, F. M.: Structure and function of the global ocean microbiome, Science, 348, 1261359, https://doi.org/10.1126/science.1261359, 2015.
Talley, L. D.: Descriptive physical oceanography: an introduction, Academic Press, https://doi.org/10.1016/C2009-0-24322-4, 2011.
Teeling, H., Fuchs, B. M., Becher, D., Klockow, C., Gardebrecht, A., Bennke, C. M., Kassabgy, M., Huang, S., Mann, A. J., Waldmann, J., and Weber, M.: Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom, Science, 336, 608–611, https://doi.org/10.1126/science.1218344, 2012.
Traving, S. J., Thygesen, U. H., Riemann, L., and Stedmon, C. A.: A model of extracellular enzymes in free-living microbes: which strategy pays off?, Appl. Environ. Microb., 81, 7385–7393, https://doi.org/10.1128/AEM.02070-15, 2015.
Vidal-Melgosa, S., Sichert, A., Francis, T. B., Bartosik, D., Niggemann, J., Wichels, A., Willats, W. G., Fuchs, B. M., Teeling, H., Becher, D., and Schweder, T.: Diatom fucan polysaccharide precipitates carbon during algal blooms. Nat. Commun., 12, 1–13, https://doi.org/10.1038/s41467-021-21009-6, 2021.
Wakeham, S. G., Lee, C., Hedges, J. I., Hernes, P. J., and Peterson, M. J.: Molecular indicators of diagenetic status in marine organic matter, Geochim. Cosmochim. Ac., 61, 5363–5369, https://doi.org/10.1016/S0016-7037(97)00312-8, 1997.
Wegner, C.-E., Richter-Heitmann, T., Klindworth, A., Klockow, C., Richter, M., Achstetter, T., Glockner, F. O., and Harder, J.: Expression of sulfatases in Rhodopirellula baltica and the diversity of sulfatases in the genus Rhodopirellula, Mar. Genom., 9, 51–61, https://doi.org/10.1016/j.margen.2012.12.001, 2013.
Weiss, M. S., Abele, U., Weckesser, J., Welte, W. U., Schiltz, E., and Schulz, G. E.: Molecular architecture and electrostatic properties of a bacterial porin, Science, 254, 1627–1630, https://doi.org/10.1126/science.1721242, 1991.
Xing, P., Hahnke, R. L., Unfried, F., Markert, S., Huang, S., Barbeyron, T., Harder, J., Becher, D., Schweder, T., Glöckner, F. O., and Amann, R. I.: Niches of two polysaccharide-degrading Polaribacter isolates from the North Sea during a spring diatom bloom, ISME J., 9, 1410, https://doi.org/10.1038/ismej.2014.225, 2015.