the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Mar 2023
| 07 Mar 2023
Abrasion of sedimentary rocks as a source of hydrogen peroxide and nutrients to subglacial ecosystems
Beatriz Gill-Olivas
Jon Telling
Mark Skidmore
Martyn Tranter
Glaciers and ice sheets are renowned for their abrasive power, yet little is known of the mechanochemical reactions which are initiated by abrasion in these environments and their effect on subglacial biogeochemistry. Here, we use sedimentary rocks representative of different subglacial environments and from a previously glaciated terrain, along with subglacial sediments, to investigate the potential for subglacial erosion to generate H2O2 and to release bio-utilisable organic carbon and nutrients (N, Fe). Samples were crushed using a ball mill, water was added to rock powders within gastight vials, and samples were incubated in the dark at 4 ∘C. Headspace and water samples were taken immediately after the addition of water and then again after 5 and 25 h. Samples generated up to 1.5 µmol H2O2 g−1. The total sulfur content, a proxy for the sulfide content, did not correlate with H2O2 generation, suggesting that the pyrite content was not the sole determinant of net H2O2 production. Other factors including the presence of carbonates, Fe-driven Fenton reactions and the pH of the solution were also likely to be important in controlling both the initial rate of production and subsequent rates of destruction of H2O2. Further, we found that erosion can provide previously unaccounted sources of bio-utilisable energy substrates and nutrients, including up to 880 nmol CH4 g−1, 680 nmol H2 g−1, volatile fatty acids (up to 1.7 µmol acetate g−1) and 8.2 to subglacial ecosystems. These results highlight the potentially important role that abrasion plays in providing nutrient and energy sources to subglacial microbial ecosystems underlain by sedimentary rocks.
- Article
(1327 KB) - Full-text XML
-
Supplement
(425 KB) - BibTeX
- EndNote





The link between physical erosion and the products of chemical weathering has long been established (Anderson, 2005). However, only a few studies have attempted to understand the chemical reactions triggered explicitly by mechanical abrasion within these environments. These studies have primarily focused on the potential reactivity of silicates after comminution (Gill-Olivas et al., 2021; Telling et al., 2015) and the release or production of gases during comminution (Gill-Olivas et al., 2021; Macdonald et al., 2018). It has been shown that comminution of certain rocks and minerals can generate strong redox agents, such as H2 (Saruwatari et al., 2004; Takehiro et al., 2011; Telling et al., 2015; Wakita et al., 1980; Edgar et al., 2022) and H2O2 (Bak et al., 2017; Borda et al., 2003; He et al., 2021; Edgar et al., 2022; Stone et al., 2022). These are expected to have an influence on the wider hydrochemistry of subglacial systems, particularly as biogeochemical reactions in these environments rely heavily on redox gradients (Tranter et al., 2005).
To date, studies of subglacial biogeochemistry have largely focused on both abiotic and biotically mediated weathering reactions (Hodson et al., 2008; Tranter et al., 2002b; Skidmore et al., 2010). Subglacial environments are often dominated by the comminution products of silicate and/or siliciclastic rocks (Wadham et al., 2010; Hodson et al., 2000). Nevertheless, it was thought until recently that their effect on the hydrochemistry of glacier systems was minimised by their slow dissolution kinetics (Lerman, 1988), with significant quantities of solute derived from silicate weathering only being associated with larger ice sheets, where there is a longer subglacial water residence time (Wadham et al., 2010). In contrast, carbonate weathering, which has more rapid dissolution kinetics, is thought to dominate the subglacial hydrochemistry even when only present in trace quantities in the comminuted bedrock (Tranter et al., 1993). This is particularly true of smaller-catchment glaciers (Wadham et al., 2010). Nevertheless, carbonate weathering is partly constrained by the availability of protons (H+; Reaction R1; Plummer et al., 1978). H+ can be derived from carbon dioxide (CO2) dissolving in water and generating carbonic acid (H2CO3; Reaction R2; Plummer et al., 1978). In subglacial systems, where atmospheric CO2 is limited, Reactions (R1) and (R2) are partly regulated by CO2 generated from organic-matter oxidation (Tranter et al., 2002a). Weathering may become dominated by carbonate hydrolysis in the absence of other proton sources, which does not require a source of H+ or CO2 (Reaction R3; Plummer et al., 1978).
Sulfide (most commonly, FeS2) oxidation is often a source of H+ in subglacial environments (S. N. Montross et al., 2013). Sulfide oxidation can be coupled to both carbonate and silicate hydrolysis (Tranter, 2003; Reactions R4 and R5, respectively).
These reactions occur when glacial flour, crushed bedrock, comes into contact with water at the glacier bed. It is often the case that trace quantities of sulfides and carbonates are liberated from the matrix of silicate minerals by crushing, but there are other effects of physical abrasion or comminution on subglacial biogeochemistry, our knowledge of which is still in its infancy. The potential solute contribution from abraded sediments of Subglacial Lake Whillans (SLW), West Antarctica, was briefly discussed in Gill-Olivas et al. (2021). These heavily weathered sediments released significant concentrations of biologically relevant solutes, such as ammonium () and acetate (CH3COO−), as well as other major ions, most likely from fluid inclusions, following comminution (Gill-Olivas et al., 2021). The abrasion of silicate minerals under simulated subglacial conditions has been shown to produce hydrogen (H2; Telling et al., 2015) through mechanochemical reactions (Reactions R6–R8; Kita et al., 1982). The solubility of H2 in water increases at low temperatures and high pressures (Wiebe and Gaddy, 1934). This makes mechanochemical reactions a potentially significant source of H2 for microbial processes, including methanogenesis in subglacial waters (Gill-Olivas et al., 2021; Telling et al., 2015; Dunham et al., 2021). Abrasion of minerals can also generate protons through the generation and dissolution of CO2 (Macdonald et al., 2018) or through the heterolytic cleavage of Si–O bonds (Reaction R9; Stillings et al., 2021), further lowering the saturation index with respect to carbonates.
Abrasion of FeS2 can also trigger chemical reactions, including Fenton reactions (Gil-Lozano et al., 2017). Crushing breaks S–S bonds in FeS2, generating dangling S− sites (Nesbitt et al., 1998). These S− sites are then stabilised by acquiring an electron from the adjoining Fe2+ ions, thus producing surface Fe3+ and S2− (Reaction R10). Water is split by the surface Fe3+ to produce ⋅OH (Reaction R11; Borda et al., 2003). Thereafter, two ⋅OH can react together to produce H2O2 (Reaction R12; Borda et al., 2001). There are multiple sources and sinks for surface and free radicals in natural systems (Gill-Olivas et al., 2021), and there is currently a need to address how the complex mineralogy of natural rock and sediment samples may hinder or enhance the relative magnitude of these reactions.
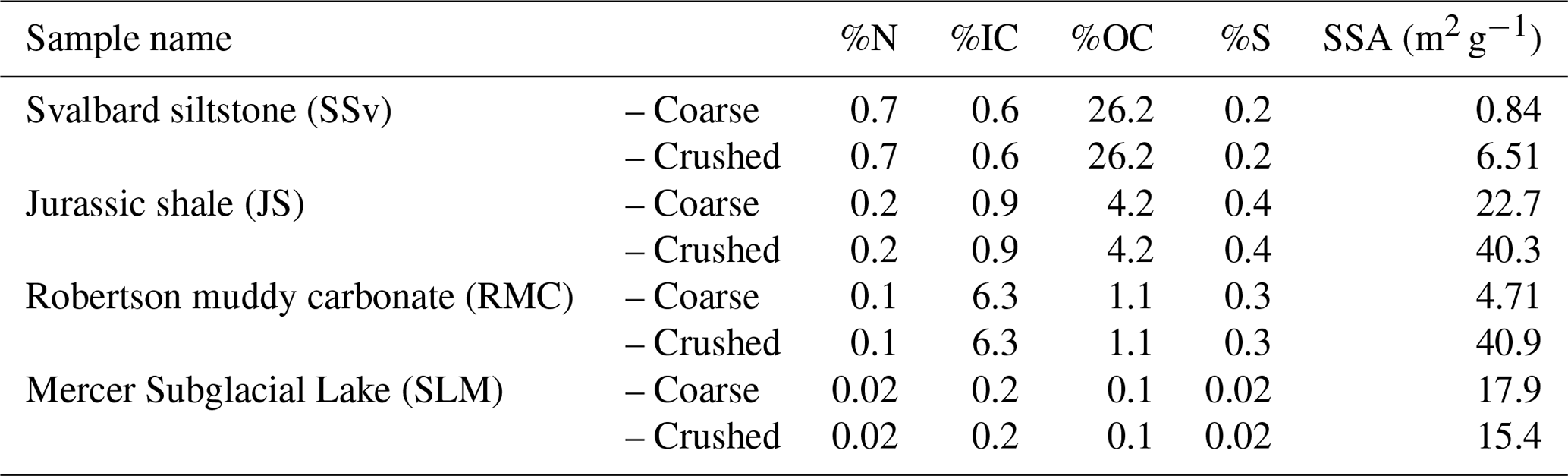
Table 1Total concentration of N, inorganic carbon (IC), organic carbon (OC) and S, as determined by elemental analysis, and specific surface area (SSA) of sedimentary rock and subglacial sediment samples. “Coarse” samples were broken up to 125 µm to 2 mm in size using a mallet and plate (JS and RMC) or a pestle and mortar (SSv and SLM). “Crushed” samples were subsampled from “coarse fractions” and ground using a planetary ball mill (500 rpm for 30 min).
The production of H2O2 by comminution may have significant consequences for the organic matter (OM) found in these environments. Subglacial environments are largely isolated from external OM inputs, but as certain glaciers form, they may override and incorporate pre-existing soil (Kohler et al., 2017), vegetation (Souchez et al., 2006) and/or lake and marine sediments (Michaud et al., 2016), thus integrating any associated OM into subglacial sediments (Wadham et al., 2019). This overridden OM can fuel microbial activity to produce CO2 or CH4 (Stibal et al., 2012). These gases are then either utilised by further microbial activity (Michaud et al., 2017), transported within the hydrological system or, in the case of CH4, potentially stored as gas hydrates (Wadham et al., 2019). Additionally, OM contains organic acids which can act as an additional proton source for subglacial weathering (S. N. Montross et al., 2013). Hydrogen peroxide is a strong oxidising agent which can oxidise OM to CO2. Thus, H2O2 production via erosion of these sediments could be problematic to microbial ecosystems if the concentration of H2O2 exceeds 0.08 % (Medina-Cordoba et al., 2018). However, it has been suggested that H2O2 might serve to oxidise and reactivate otherwise refractive organic molecules in subglacial systems (Tranter, 2015). A number of studies have concluded that DOC (dissolved organic carbon) and POC (particulate organic carbon) exported from glacial systems is highly bio-available (Lawson et al., 2014), yet is also paradoxically ancient (Hood et al., 2009), lending some credence to the potential re-activation of ancient OM through erosion (Tranter, 2015).
Here, we aim to further understand the generation of H2O2 by the comminution of various pyrite-containing sedimentary rocks and subglacial sediments. We hypothesise that the concentration of sulfide in sedimentary rocks would have a first-order impact on the amount of H2O2 that is generated when the rocks are crushed. Further, we aim to investigate how the generation of H2O2 may affect the availability of OM present in these rocks and sediments and the potential release of bio-available compounds. We do this by investigating the H2O2 generation, the overall water chemistry and the release of gases when three different sedimentary rocks (shale, siltstone and muddy carbonate samples) and one subglacial sediment sample, chosen for their varying C, OC and S contents (Table 1), were crushed and incubated with water at 4 ∘C. The experimental design for this study is consistent with previous subglacial-abrasion studies. These experiments are not perfect analogues of subglacial environments, being conducted at slightly warmer temperatures and lower pressures than those of natural systems, but they illustrate the potential contribution of subglacial abrasion and wetting of crushed sediments to the chemical weathering and provision of chemicals that occur in biogeochemical reactions in these environments. We elaborate on these issues in the discussion.
2.1 Sample sites
A selection of rocks and a sediment sample spanning a range of carbon (C), nitrogen (N), sulfur (S) and organic carbon (OC) from three different glaciated catchments and a rock sampled from a previously glaciated terrain were utilised in this study. Jurassic shale (JS) from Yorkshire (UK), purchased from Northern Geological Supplies Ltd., was used as a positive control for these experiments, as it is known to be relatively high in pyrite and has an organic-carbon content that is in the middle range of the sample types chosen. Further, these rocks were likely overlain by ice during the Last Glacial Maximum. Samples from glaciated environments included rock samples collected near Longyearbreen (SSv; Svalbard, 78.1941∘ N, 15.5475∘ W) and Robertson Glacier (RMC; Canadian Rockies, 50.738200∘ N, 115.331734∘ W) and a subglacial sediment sample from Mercer Subglacial Lake (SLM; Antarctica, (84.640287∘ S, 149.501340∘ W). The bedrock underlying Longyearbreen and surrounding areas are primarily from the Early Cretaceous Epoch (Helvetiafjell and Carolinefjell formations) and from the Late Palaeocene to Early Eocene epochs (Firkanten, Basilika and Battfjellet formations) and consist primarily of sedimentary rocks including sandstones, siltstones, mudstones and shales (Elvevold et al., 2007). The local bedrock underlying Robertson Glacier is Upper Devonian in age (Mount Hawk, Palliser, and Sassenach formations) and consists of impure limestones, dolostones and dolomitic limestones, with interbeds of shale, siltstone and sandstone (McMechan, 1998). The sample from SLM was material from the core catcher from the base of the sediment gravity corer (SLM1801-02FF-2) at a sediment depth 2.06 m (Priscu et al., 2021). The basal sediments from the core were clast-rich, massive, muddy glacial diamicts (Campbell et al., 2019), similar in composition to the diamicts from beneath the neighbouring Whillans Ice Stream (Tulaczyk et al., 1998) and Subglacial Lake Whillans (SLW) (Hodson et al., 2016). The matrix-supported diamict was very poorly sorted (Folk and Ward, 1957), with the matrix being composed of predominantly silt (52 %) with contributions of sand (25 %) and clay (23 %) (Campbell et al., 2019).
2.2 Elemental analysis
C, N and S concentrations in the samples were determined using an elemental analyser (Elementar Vario PYRO Cube, Langenselbold, Hesse, Germany). Triplicate 5 to 20 mg of crushed samples were weighed into tin capsules and introduced into the machine. The organic carbon (OC) concentrations were determined following techniques in Harris et al. (2001). Some 20 to 30 mg of crushed samples was weighed into silver capsules (part no. S05003397). The capsules were transferred to a microtiter plate and wetted with 50 µL of 18.2 MΩ water, and then they were transferred to a desiccator. A beaker with ∼ 100 mL 12 M HCl was also placed inside this desiccator, and the samples were exposed to the HCl vapours overnight (∼ 14 h), dried in an oven at 60 ∘C for 4 h, and then placed within another silver capsule and closed up. These fumigated samples were then introduced into the elemental analyser (Elementar Vario PYRO Cube, Langenselbold, Hesse, Germany) as before. The detection limit was 0.001 % for all three elements, and the coefficients of variation (CV) for C, N and S for eight replicates of a soil analytical standard (NCS Soil Standard 338 40025, cert. 133317; C = 2.29 %, N = 0.21 %, S = 0.031 %; Elemental Microanalysis Ltd., United Kingdom) were 4.9 %, 5.3 % and 18 %, respectively.
2.3 Sample preparation and dry crushing
The rock samples were washed with 18.2 MΩ cm−1 water and dried at 75 ∘C for over 18 h (after which there was no mass change). RMC and JS samples were then wrapped in several layers of paper roll bags (to avoid metal–rock interaction) and broken using a sledgehammer on a metal plate. SSv was broken using an agate pestle and mortar, and SLM sediments were disaggregated also using an agate pestle and mortar. The broken rocks and sediment were sieved, and the 125 µm to 2 mm size fraction was collected (hereafter referred to as the “coarse” samples). The coarse samples were then washed with 18.2 MΩ cm−1 water, transferred to a foil tray and covered with a loosely fitted foil cover, and dried at 75 ∘C overnight or until there was no change in mass. The “crushed” samples were prepared by crushing 15 g of the clean and dried coarse samples in air for 30 min within a zirconium oxide ball mill at 500 rpm using a FRITSCH Planetary Mono Mill PULVERISETTE 6 (FRITSCH GmbH, Idar-Oberstein, Germany). After crushing, the size of rock and sediment samples ranged from 0.3 to 806 µm (measured using a Malvern Mastersizer 3000 laser particle size analyser; Malvern Panalytical, Worcestershire, UK).
2.4 Microcosm experiments
Triplicate 1 g coarse samples for each time point were weighed into 50 mL borosilicate serum vials (Wheaton®, VWR), which had been previously acid washed, rinsed six times with 18.2 MΩ cm−1 water and then maintained at 450 ∘C for 4 h in a muffle furnace. These vials were then sealed with grey butyl rubber stoppers, which had been previously washed in 3 % Decon® 90, and were then acid washed, soaked in 18.2 MΩ cm−1 water overnight before being rinsed six times with 18.2 MΩ cm−1 water, autoclaved and dried. The same procedure was followed for crushed samples immediately after milling. Three borosilicate vials were sealed without the addition of samples for use as rock-free controls (blanks) at each time point. Then, 15 mL aliquots of 18.2 MΩ cm−1 water, which had been pre-autoclaved and cooled to 4 ∘C, were added to each vial in turn. The vials were shaken by hand to mix, and a 10 mL headspace gas sample was taken by over-pressurising the vials with 10 mL of N2 and transferring the gas into a 5.9 mL Exetainer® (Labco, Lampeter, UK). Next, the serum vials were opened, and the slurry was filtered using a 0.22 µm PES Steriflip® vacuum filtration system (Millipore). A 4 mL filtrate aliquot was taken to analyse pH (using Hach® Sension+ 5208) followed by analysis of H2O2 concentration. The same procedure was followed for samples taken 5 and 25 h after the addition of water.
2.5 Hydrogen peroxide analysis
A 4 mL aliquot of the filtered solution was used for H2O2 analysis using neocuprine (2,9-dimethyl-1,10-phenanthroline (product no. N1501; Sigma-Aldrich)), based on the methods in Baga et al. (1988) and Borda et al. (2001). Standards (0 to 100 µM) were prepared using known concentrations of H2O2 (ESMURE® ISO; 30 % hydrogen peroxide, Perhydrol®; Millipore); 4 mL of each standard was pipetted into a 15 mL centrifuge tube, and to this, 1 mL of 0.01 M copper (II) sulfate and 1 mL of neocuprine solution (10 g L−1 in ethanol) were added. The solution was then made up to 10 mL with 18.2 MΩ water, and the absorbance of the solution was measured at 454 nm using a Shimadzu UVmini 1240 UV–VIS spectrophotometer (Shimadzu, Kyoto, Japan).
2.6 Gas analysis
Headspace gas samples were taken from the ball mill before and after crushing by over-pressurising the ball mill with 10 mL of oxygen-free N2 (zero grade, BOC). Then, a 10 mL headspace gas aliquot was transferred into a 5.9 mL pre-evacuated double-waded Exetainer® (Labco, Lampeter, UK) using a gas-tight syringe. Headspace gases from the vials were sampled immediately after the addition of water and after 5 and 25 h. The contents of the Exetainers® were then analysed using an Agilent 8860 Gas Chromatograph (Agilent Technologies, Santa Clara, CA, USA). Concentrations of CH4 and CO2 were determined by a flame ionisation detector (FID). The sample loop was 0.5 mL, helium (He) was used as the carrier gas, and a Porapak Q 80–100 mesh, 2 m × 3.8 mm × 2 mm SS column and a methaniser were used to distinguish the compounds. The concentrations of H2 and O2 were determined using a thermal conductivity detector (TCD), a 1 mL sample loop, argon (Ar) as the carrier gas and a Hayesep D 80–100 mesh, 2 m × 3.18 mm ( in.) SS column in series with a molecular sieve 5a, 60–80 mesh, 1.52 m × 3.18 mm (8 ft × in.) column. The oven temperature was set at 30 ∘C for the initial 4 min, and then the temperature was ramped up at rate of 50 until the oven reached a temperature of 200 ∘C. This temperature was maintained for 2.5 min, when the run was concluded. The concentrations of headspace gases were calculated based on a standard curve generated from the dilution of an 11-mixed-gas standard (173738-AH-C, BOC). The standard curve was linear over the concentration ranges for H2 (4.1–502 ppm, R2 = 0.9988, n = 6), for CH4 (1.6–198 ppm, R2 = 0.9988, n = 7) and for CO2 (3.4–398 ppm, R2 = 0.9992, n = 7). Standards were run daily and gave a CV of 1.2 % (n = 31) for H2, with a detection limit of 2.2 ppm, equivalent to 5.2 nmol g−1; a CV of 1.2 % (n = 31) for CH4, with a detection limit of 0.11 ppm, equivalent to 0.26 nmol g−1; and a CV of 4.3 % (n = 31) for CO2, with a detection limit of 0.40 ppm, equivalent to 0.93 nmol g−1. The ideal gas law was used to convert to molar concentrations, and concentrations were corrected for dilution during sampling and for gases dissolved in the water using Henry's diffusion coefficient (where relevant). The results were also blank corrected and normalised to dry-sediment mass.
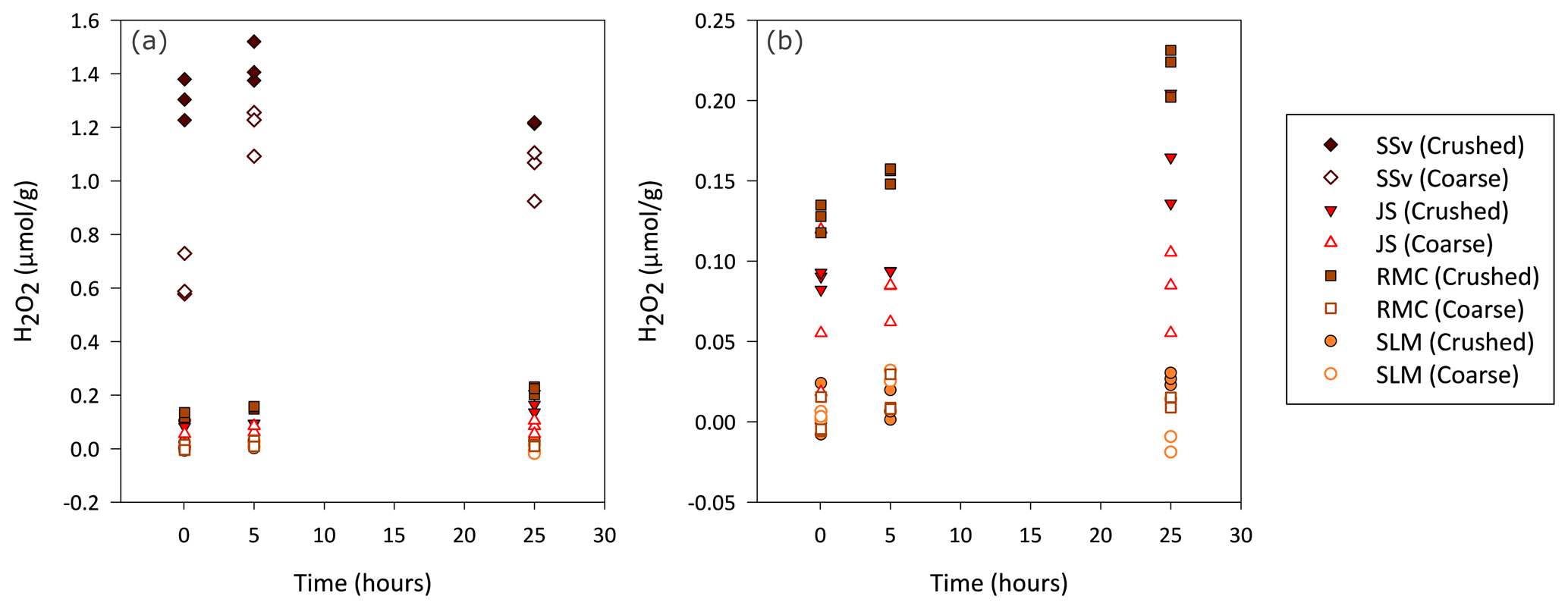
Figure 1Concentration of H2O2 generated by coarse and crushed sedimentary rock (SSv, JS and RMC) and subglacial sediment samples (SLM). (a) Blank-corrected H2O2 (µ mol) produced per gram of crushed and coarse samples during a 25 h aerobic incubation of these samples with water. (b) Focus on lower concentrations of H2O2, excluding results of coarse and crushed SSv samples, to allow a clearer view of the data distribution of the remaining samples.
2.7 Water chemistry analysis
Anions and organic acids including acetate (limit of detection (LOD): 0.052 ppm, equivalent to 0.14 µmol g−1; CV: 0.76 %), formate (LOD: 0.018 ppm, equivalent to 0.06 µmol g−1; CV: 1.07 %), (LOD: 0.22 ppm, equivalent to 0.37 µmol g−1; CV: 1.41 %) and oxalate (LOD: 0.020 ppm, equivalent to 0.04 µmol g−1; CV: 0.48 %) were analysed using a Dionex ICS 6000 (Thermo Scientific), fitted with a Dionex IonPac™ AS11-HC 4 µm column.
Additional aliquots of the filtered solution were subsampled to analyse for (LOD: 0.026 ppm, equivalent to 0.3 µmol g−1; CV: 2.3 %) and total Fe (LOD: 0.17 ppm, equivalent to 0.5 nmol g−1; CV: 1.5 %) using the Gallery Automated Photometric Analyzer (Thermo Fisher). For analysis, aliquots of JS and SLM were diluted to 1 in 10, and aliquots of SSv and RMC were diluted to 1 in 20 (due to their concentration being greater than the highest standard). Concentrations of were then determined using an adapted version of the salicylate method outlined in Le and Boyd (2012). The concentration of total Fe was determined using an adapted version of the ferrozine method described in Viollier et al. (2000) using undiluted aliquots of the filtered solution.
2.8 Specific surface area
The specific surface area (SSA) of materials was measured using a NOVA 1200e BET analysis system. Approximately 1 g of dried sample was loaded into a pre-calibrated sample cell. The whole system was then evacuated and dried at 110 ∘C for a minimum of 6 h (Zhou et al., 2019). The surface area of materials was measured using nitrogen gas as an adsorbent at 77 K.
2.9 Statistical analysis
Simple regression lines were fitted to infer potential correlations between the concentration of H2O2 produced by crushed and coarse samples and sulfate, iron or specific surface area using SigmaPlot. An R2 value of 0.7 or above was considered to indicate a strong correlation between two variables (Moore et al., 2013). A Kruskal–Wallis test was conducted followed by a post hoc Wilcoxon test using RStudio to determine significant differences in the concentrations of , acetate, formate and pH between crushed and coarse samples. The data for these compounds and pH did not show a temporal trend; thus the values were pooled into one population, and the Kruskal–Wallis test determined that the distribution of the data was non-parametric. Due to this and the low number of samples, a Wilcoxon test was then used to determine statistical differences in the concentrations of these compounds and pH in crushed and coarse samples.
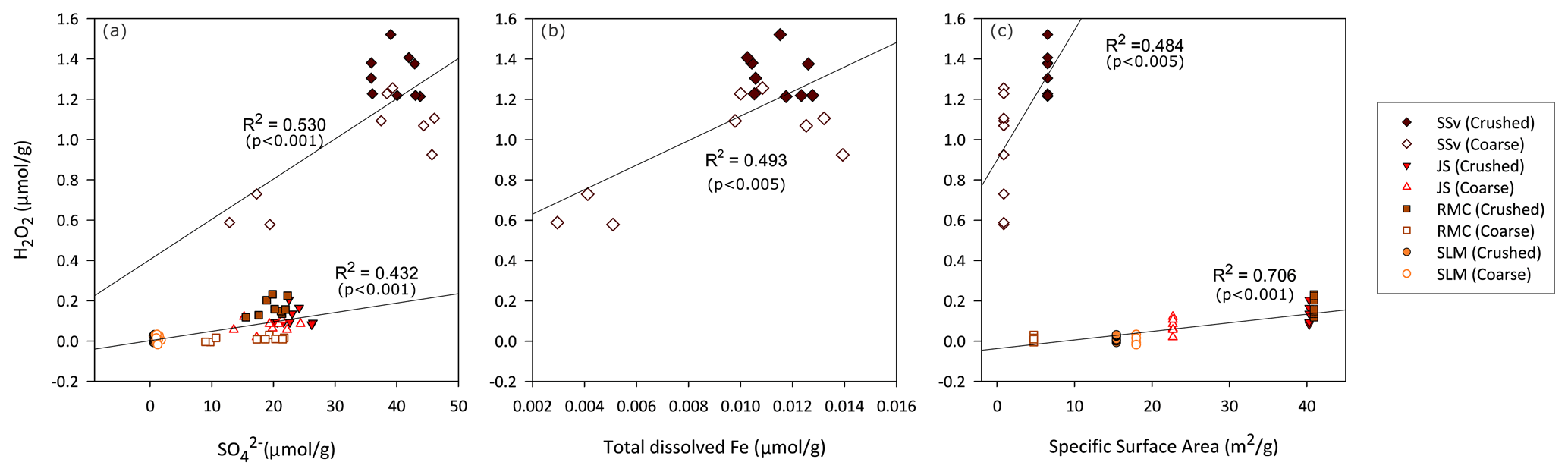
Figure 2Relationships between concentration of H2O2 (µ mol) produced per gram of crushed and coarse rock and sediment samples relative to (a) sulfate in solution, (b) iron in solution and (c) specific surface area of samples. The concentrations of H2O2, sulfate (a) and dissolved Fe (b) measured in solution are normalised to gram of sample and treated as one population throughout the 25 h of incubation. Note that Fe concentrations were above detection limits only in the SSv samples. R2 and p values obtained from linear regression model in RStudio.
3.1 Hydrogen peroxide
Overall, crushed samples produced more H2O2 than their coarse counterparts, and the concentration increased over the first 25 h. SSv samples produced the highest concentration of H2O2, with crushed samples producing up to 1.5 µmol H2O2 g−1 (Fig. 1a) and coarse samples producing up to 1.3 µmol H2O2 g−1 (Fig. 1a). The concentration of H2O2 in SSv increased between the 5 min and 5 h time points but then decreased at the 25 h time point in contrast to the continual rise in concentration shown by the other samples. The crushed RMC and JS samples, which also produced quantifiable H2O2 concentrations, generated an order of magnitude less H2O2 than crushed SSv samples, with maximum concentrations of 0.23 and 0.20 µmol g−1, respectively (Fig. 1b). The concentration of H2O2 produced by coarse JS samples was only slightly lower than that of its crushed counterpart (maximum of 0.12 µmol g−1; Fig. 1b). The concentration produced by coarse RMC was considerably lower than its crushed equivalent, reaching a maximum of 0.02 µmol g−1 (Fig. 1b). SLM samples barely produced any H2O2 above the LOD; only after 25 h was the concentration of H2O2 in crushed samples quantifiable (maximum value of 0.03 µmol g−1; Fig. 1b). This contrasts with the concentrations of H2O2 produced by crushed Subglacial Lake Whillans samples, which were much higher (up to 17 µmol g−1; Gill-Olivas et al., 2021).
Production of H2O2 was weakly correlated to the concentration of in the solution in samples with low H2O2 (R2 = 0.432; Fig. 2a) and more strongly correlated with the SSA of these samples (R2 = 0.706; Fig. 2c). The concentration of H2O2 produced by SSv did not fall on these regression lines. The production of H2O2 from SSv samples shows a weak correlation with and SSA (R2 = 0.530 – Fig. 2a; R2 = 0.484 – Fig. 2c); however, this is a limited sample size, so care should be taken in interpreting these correlations. Total dissolved Fe was only detected in SSv samples, and this also showed a weak correlation with H2O2 concentrations (R2 = 0.493; Fig. 2b).
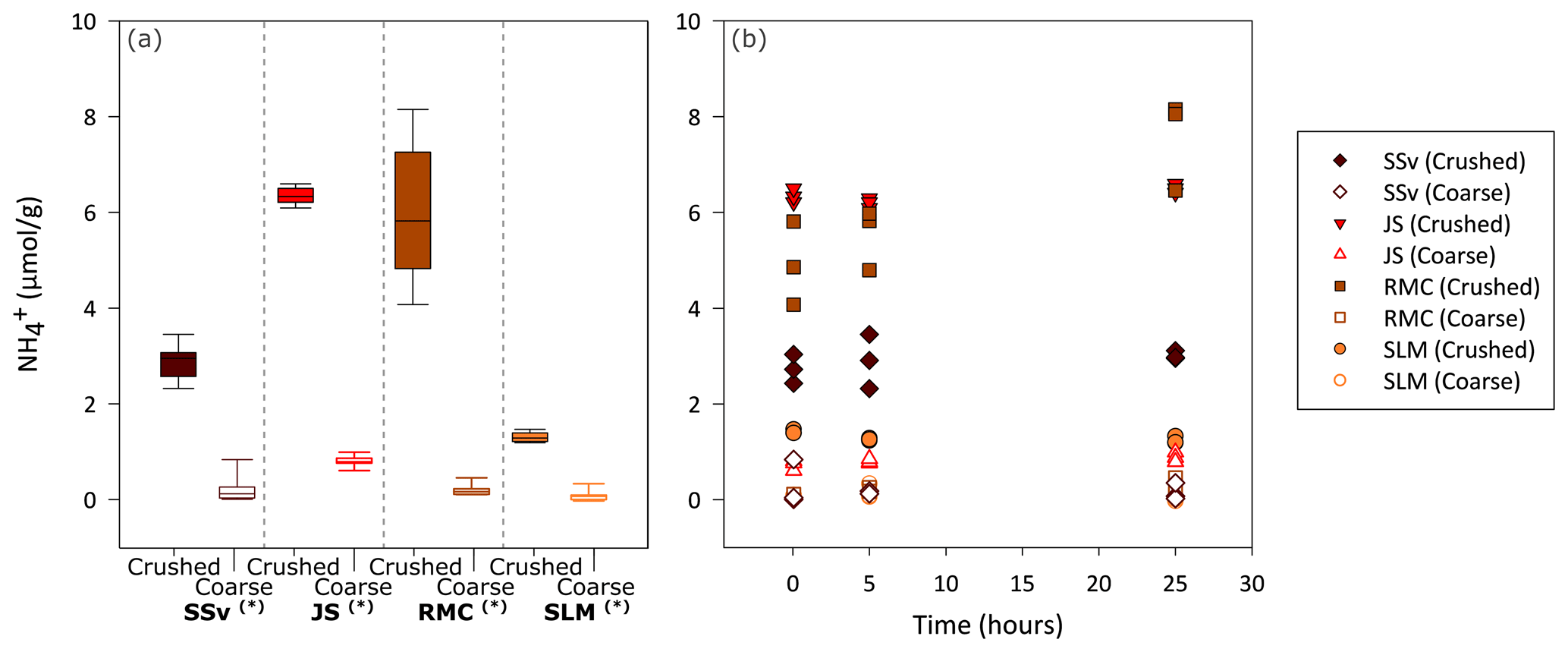
Figure 3Concentration of in solution during 25 h incubation of crushed and coarse rock and sediment samples. (a) Boxplot showing the range of measured in solution (normalised to ) for all time points (5 min, 5 h and 25 h) in the incubation of coarse and crushed samples (n = 9 for each sample type). Highly significant difference (p ≤ 0.01) between crushed and coarse samples is denoted by (∗). The minimum and maximum values are represented by the whiskers, the interquartile range is represented by the box, and the median value is shown by the line in the centre of the box. (b) Concentration of in solution (normalised to ) after approximately 5 min and 5 h and 25 h of incubation.
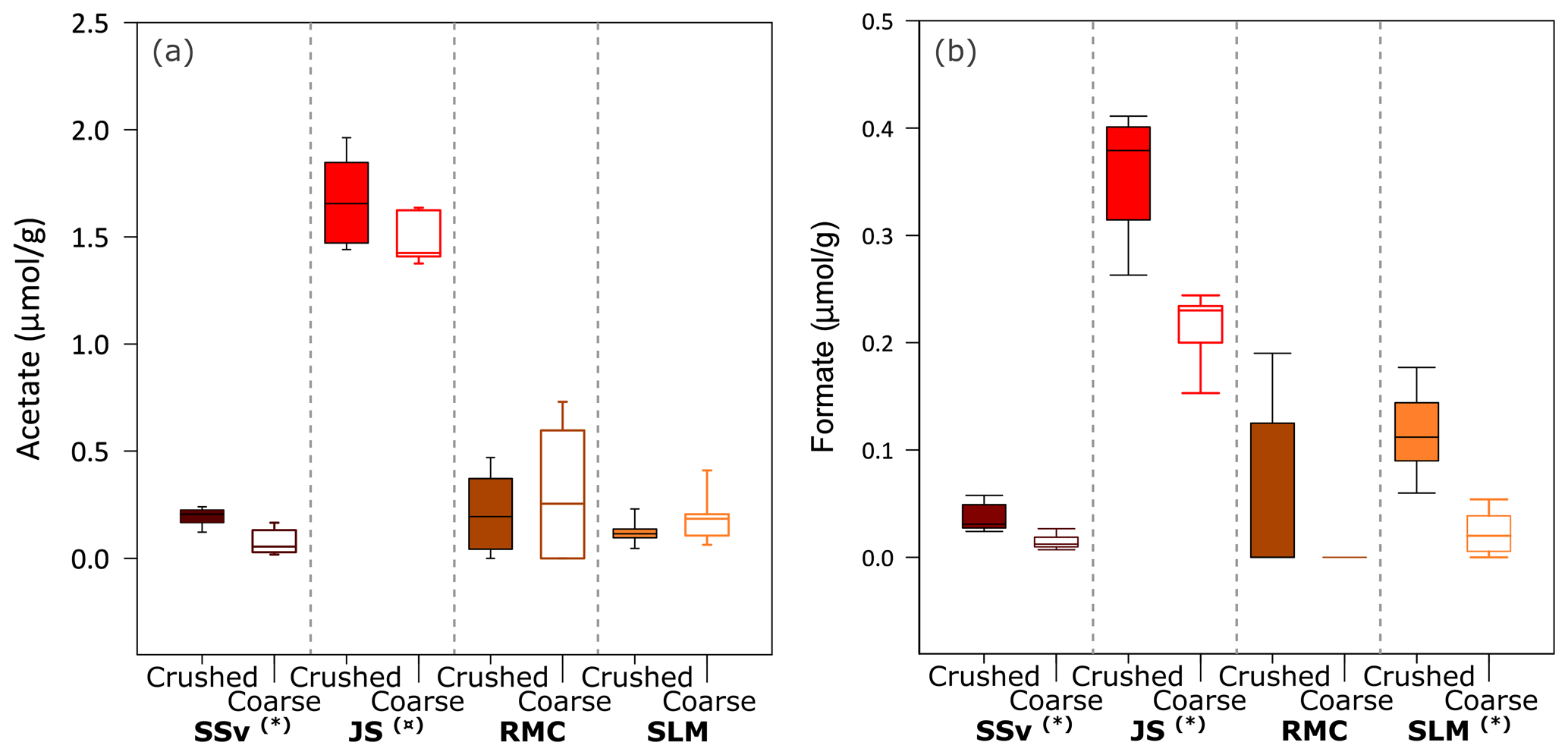
Figure 4Concentration of acetate and formate in solution from incubation of crushed and coarse rock and sediment samples with ultrapure water at all time points (5 min, 5 h and 25 h). Boxplots of (a) acetate concentration in solution (normalised to µmol acetate g−1) and (b) formate concentration in solution (normalised to µmol formate g−1) after the incubation of coarse and crushed samples (n = 9 for each sample type). Highly significant difference (p ≤ 0.01) between crushed and coarse samples is denoted by (∗). Significant difference (p ≤ 0.05) between crushed and coarse samples is denoted by (![]() ). The minimum and maximum values are represented by the whiskers, the interquartile range is represented by the box, and the median value is shown by the line in the centre of the box.
). The minimum and maximum values are represented by the whiskers, the interquartile range is represented by the box, and the median value is shown by the line in the centre of the box.
3.2 Ammonium in solution
All crushed samples produced significantly higher concentrations of in solution compared to their coarse counterparts (Wilcoxon test, p ≤ 0.01; Fig. 3a). There was no clear temporal trend to the production of other than, potentially, a slight increase with time for RMC samples (Fig. 3b). After 25 h, crushed RMC samples showed the highest concentrations (up to 8.2 µmol g−1), closely followed by crushed JS samples (up to 6.6 µmol g−1). This is considerably higher than their coarse counterparts, with concentrations of up to 0.20 and 0.99 µmol g−1 for RMC and JS samples, respectively (Fig. 3a).

Figure 5The pH of solutions at all time points (5 min, 5 h and 25 h) of the incubation of crushed and coarse rock and sediment samples with ultra-pure water (n = 9 for each sample and treatment). Highly significant difference (p ≤ 0.01) between crushed and coarse samples is denoted by (∗). The minimum and maximum values are represented by the whiskers, the interquartile range is represented by the box, and the median value is shown by the line in the centre of the box.
3.3 Organic acids
All crushed and coarse samples produced quantifiable concentrations of acetate (Fig. 4a) and, in some cases, formate (Fig. 4b). There was a significant range of values for acetate concentrations and no clear temporal trend throughout the 25 h incubations. However, SSv and JS samples showed a highly significant (Wilcoxon test, p ≤ 0.01) and significant (Wilcoxon test, p ≤ 0.05) difference between crushed and coarse samples (Fig. 4a). Formate concentrations were lower than acetate, but all samples, except for RMC, showed a highly significant difference between crushed and coarse samples (Wilcoxon test, p ≤ 0.01). JS produced the highest concentrations of both acetate (median of 1.7 µmol g−1 for crushed samples; Fig. 4a) and formate (median of 0.38 µmol g−1 crushed samples; Fig. 4b). SLM and SSv both produced considerably less acetate (median of 0.11 and 0.21 µmol g−1 for crushed samples, respectively; Fig. 4a). Formate concentrations were also low in SSv samples, and SLM samples produced slightly more formate (median of 0.03 and 0.11 µmol g−1, respectively, Fig. 4b). RMC samples showed considerable variability in acetate concentrations and little, if any, formate.
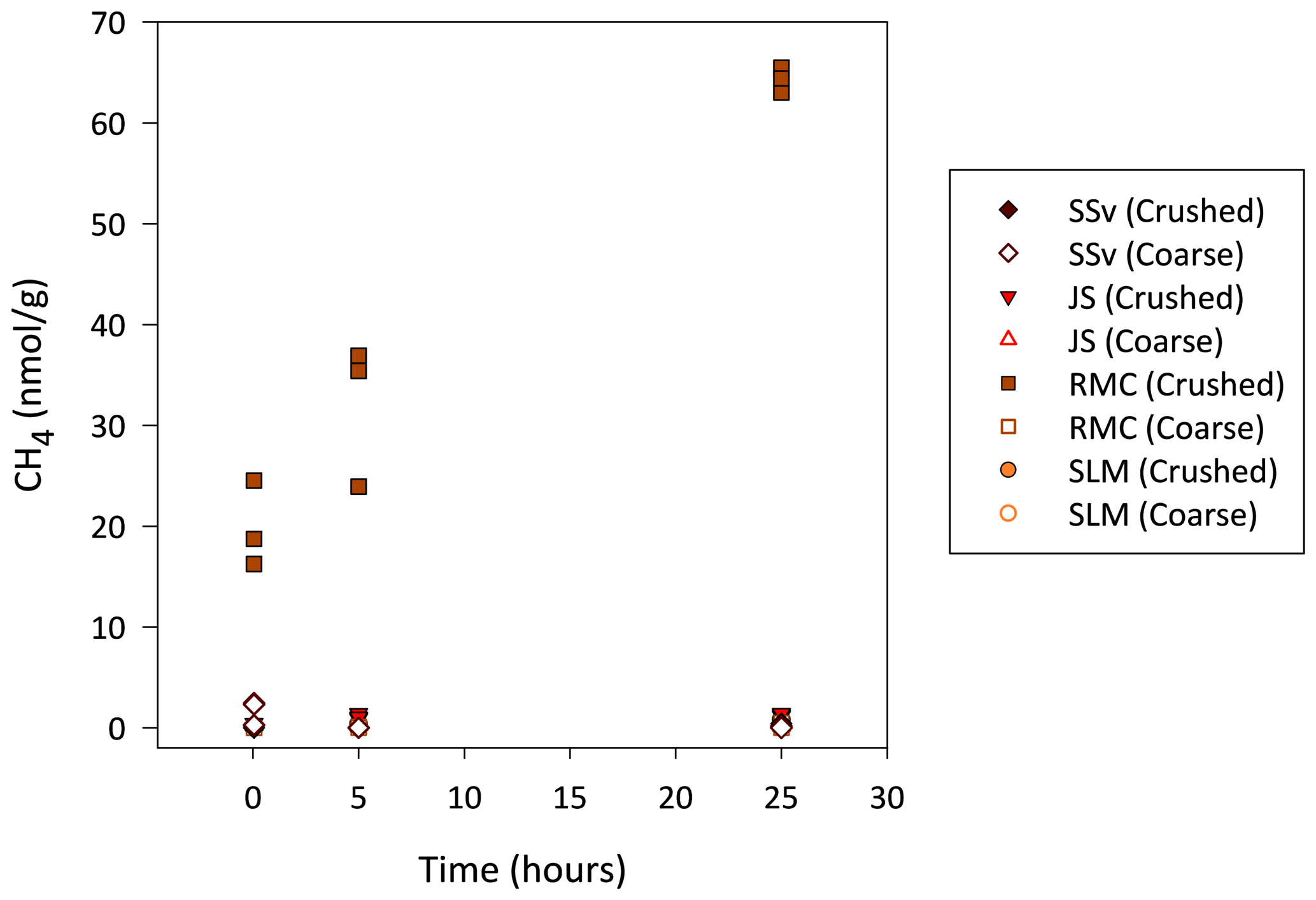
Figure 6Generation of CH4 during incubation of coarse (63 µm–2 mm) and crushed sedimentary rock and subglacial sediment samples. Headspace gas samples were analysed after 5 min, 5 h and 25 h of incubation, blank corrected and normalised to the grams of sample.
3.4 pH
Overall, the pH of all samples stayed relatively stable over the incubation time of 25 h. Crushed samples appeared to have slightly higher pH than their coarse counterparts, potentially due to the release of calcite leading to carbonate reactions, elevating the pH (Fig. 5). Most samples had pH close to neutral or above; the only exception was SSv incubations which were acidic, with a median pH of 3.61 and 3.11 in crushed and coarse samples, respectively (Fig. 5).
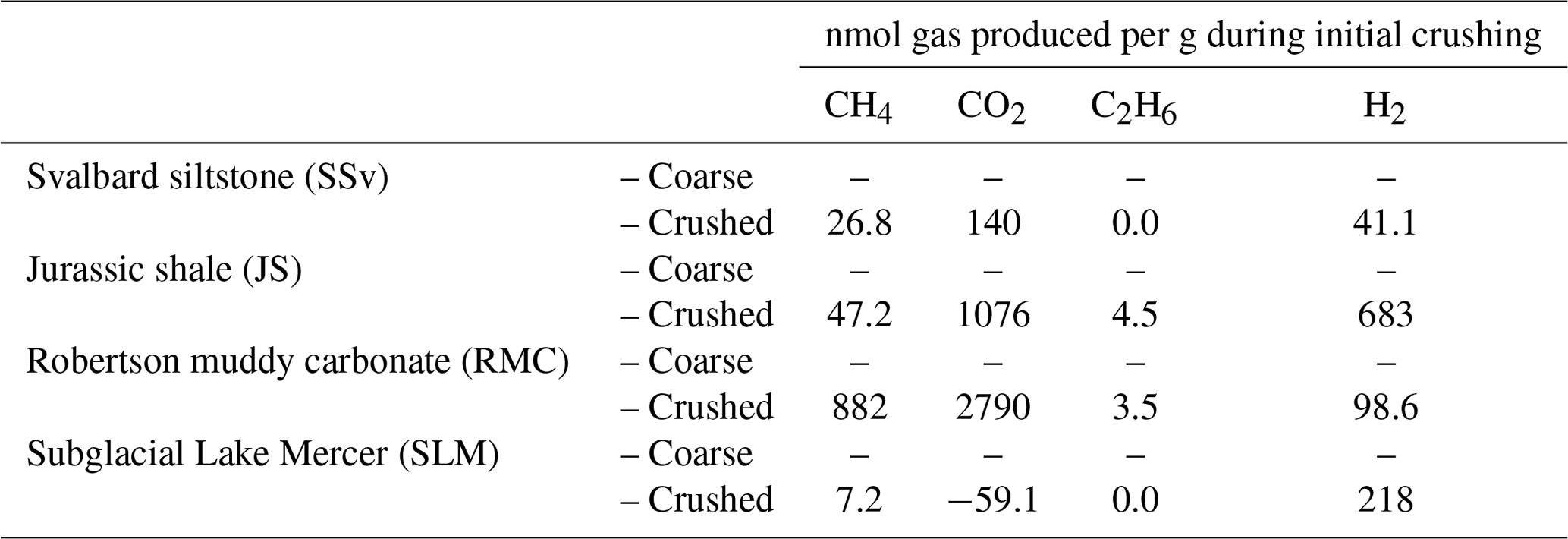
Table 2Gases released to ball mill headspace during high-energy crushing (500 rpm for 30 min) of sedimentary rock (SSv, JS and RMC) and subglacial sediment (SLM) samples. Crushing was conducted under air; therefore, values shown in this table have been corrected for the initial concentration of atmospheric gases in the ball mill headspace.
3.5 Gases released
All rock samples produced quantifiable concentrations of CH4, CO2 and H2 gas above atmospheric concentrations (Table 2). RMC produced the highest concentrations of CH4 and CO2 (880 nmol CH4 g−1 and 2800 nmol CO2 g−1; Table 2) during crushing and produced quantifiable C2H6 concentrations (3.5 nmol g−1; Table 2). JS samples produced noticeable concentrations of CH4, CO2 and C2H6 (47, 1100 and 4.5 nmol g−1, respectively; Table 2). JS samples also produced the highest H2 concentrations measured (680 nmol g−1; Table 2). SSv produced less CH4, CO2, C2H6 and H2 than JS and RMC samples, and SLM produced the lowest concentration of CH4 out of these samples (7.2 nmol g−1; Table 2) and no CO2 and C2H6 above the LOD. However, they did release significant concentrations of H2 during crushing (220 nmol g−1; Table 2).
During incubations, the concentrations of the gases CO2, H2 and C2H6 were, for the most part, indistinguishable from the noise in the analysis of these gas concentrations in laboratory air. However, crushed RMC samples did produce CH4 concentrations over the 25 h incubation which could be quantified, increasing from an average of 20 to 64 nmol g−1 (Fig. 6). JS crushed samples also produced quantifiable CH4, although at much lower concentrations, below LOD at 5 min and an average of 1.1 and 1.3 nmol g−1 after 5 and 25 h, respectively (Fig. 6).
4.1 The effect of mineralogy on oxidant production
The concentration of S in the samples (Table 1) was assumed to be indicative of pyrite content, since no other sulfur-containing minerals (e.g. anhydrite, gypsum) have been reported for the rocks or sediments (McMechan, 1998; Campbell et al., 2019; Elvevold et al., 2007). Thus, it was expected that samples with higher S content would also produce more H2O2 when crushed if crushed samples had similar surface areas. However, our results showed that SSv samples produced considerably more H2O2 (Fig. 1a) than other rock samples, despite the relatively low S content and SSA (Table 1), indicating that other factors played a role in the generation and/or consumption of H2O2. We focus on generation first.
The rupture of Fe–S and S–S bonds in pyrite has been shown to produce H2O2 in both oxic (Nesbitt et al., 1998) and anoxic conditions (Reactions R10–R12; Borda et al., 2003). Simultaneously, the release of Fe2+ into solution due to dissolution of FeS2 can catalyse the Fenton and Haber–Weiss reaction, removing H2O2 and generating other reactive oxygen species (ROS) (Reactions R13–R16; Gil-Lozano et al., 2017). Hydroxyl radicals (⋅OH) are often suggested as an intermediate, which can react together to regenerate H2O2 (Reaction R17). However, this only occurs under acidic conditions (Bataineh et al., 2012). Solutions from SSv samples had an acidic pH (Fig. 5), and so these samples may have generated more ⋅OH and could thus sustain H2O2 concentrations generated by crushing (Fig. 1a).
Fe(IV) is preferentially produced as a Fenton intermediate at circumneutral pH (Bataineh et al., 2012), thus rapidly utilising any H2O2 produced from abrasion of pyrite. Further, studies into the Fenton reaction in biological systems suggest that where is present, carbonate–radical anions () will be the most likely product of the Fenton reaction (Illes et al., 2019). Solutions incubated with JS and RMC samples had a circumneutral to basic pH (Fig. 5), and the rocks had relatively high carbonate contents (Table 1). Therefore, it is likely that was produced as a Fenton product despite the higher pyrite content in these samples, thus yielding less H2O2. The SLM samples produced limited H2O2 (Fig. 1a) despite the lower carbonate content of the sediments (Table 1). This may be due to the much lower S content of these samples (Table 1) generating lower concentrations of initial H2O2 combined with the basic pH of the solution (Fig. 5).
SSA is (strongly) correlated with H2O2 production in samples with non-acidic pH and similar S content (R2 = 0.706; Fig. 2c). It has been previously suggested that grain size (and thus SSA) is a primary control on the rate of pyrite oxidation (Rimstidt and Vaughan, 2003), and surface area has also been shown to play a key role in the kinetics of degradation of H2O2 by the Fenton reaction (Gil-Lozano et al., 2014).
4.2 Release of OC sources to the subglacial system
Microbes within subglacial environments are capable of recycling OM (e.g. Wadham et al., 2019; Lanoil et al., 2009; Priscu, 2008; Christner et al., 2014; Skidmore, 2011), and concentrations above 0.08 % (24 mM) H2O2 could inhibit these reactions by decreasing microbial populations (Medina-Cordoba et al., 2018) or by oxidising OM. Conversely, the generated H2O2 could have a net positive effect in these ecosystems by potentially re-activating legacy OM into more labile forms (Tranter, 2014). Previous crushing experiments found a significant increase in acetate between crushed and coarse sediment samples from Subglacial Lake Whillans (Gill-Olivas et al., 2021), which would provide a carbon source for both aerobic and anaerobic metabolisms, depending on redox conditions. All samples used in these experiments produced quantifiable concentrations of acetate (Fig. 4a), but only SSv and JS samples showed statistically significant difference between crushed and coarse samples (Fig. 4a). There was no temporal trend or apparent correlation with H2O2 production, SSA or %OC content of samples (Fig. S1 in Supplement A). It is worth noting that there are several methodological differences between these and previous studies, one of the most significant being the incubation time. These samples were only incubated for 25 h, whilst previous studies which showed major differences in acetate production between crushed and coarse samples were conducted over 41 d (Gill-Olivas et al., 2021). Further, the current study was conducted at 4 ∘C, a slightly higher incubation temperature than that of the previous study, 0 ∘C (Gill-Olivas et al., 2021). This temperature difference may have a minor effect on the rate of acetate production, with in situ rates closer to those at 0 ∘C.
There were no discernible temporal trends or correlations between formate and H2O2 production, SSA or %OC content of samples, but there was a noticeable increase in formate concentrations in crushed samples versus their uncrushed counterparts, even in these short incubations (Fig. 4b). There was no formate detected in previous incubations of crushed SLW samples (Gill-Olivas et al., 2021). Formate is a biologically significant compound, particularly in anaerobic environments where it has been shown to be utilised as an electron donor in a range of metabolisms (e.g. Madigan et al., 2020).
The lack of a temporal trend in organic acid concentrations would suggest that H2O2 did not affect the increase or decrease of organics. SSv had the highest OC content (Table 1), yet it had some of the lowest concentrations of acetate and formate in both crushed and coarse samples (Fig. 4). Conversely, JS and SLM samples, which produced much lower H2O2 concentrations, had much higher concentrations of these short-chain, bio-available organics despite having considerably lower %OC. Furthermore, the increase in formate between coarse and crushed samples was much more noticeable for these samples, where H2O2 concentrations were lower. These results could suggest that, while abrasion may increase the production of bio-available OC in solutions, they may be oxidised when H2O2 concentrations are also generated.
4.3 Bio-available gases
Methanogens have been found in many subglacial systems, including glaciers on Svalbard (Stibal et al., 2012; Kaštovská et al., 2007), Robertson Glacier (Boyd et al., 2010) and SLW (Michaud et al., 2017), which is within the same broad hydrological system as SLM (Carter et al., 2013). It is likely that subglacial methanogens are capable of hydrogenotrophy, utilising H2 and CO2 to generate CH4 (Stibal et al., 2012). Thus, the release of H2 in all samples and of CO2 from most samples during crushing has the potential to stimulate methanogenic populations found in these environments. Further, there have been reports of methanotrophic organisms in certain subglacial environments (Michaud et al., 2017) which could utilise the CH4 released by all crushed samples (Table 2).
Thermal degradation of OM during the formation of shales can generate H2, CO2 and CH4 which can then be trapped within pore spaces of these and associated sedimentary rocks (Suzuki et al., 2017). Erosion of these sedimentary rocks opens interpore spaces and releases any gases trapped in them (Macdonald et al., 2018). The CH4, C2H6 and CO2 released by crushing RMC samples were within the range found in previous crushing studies using muddy carbonate rock samples from Robertson Glacier (Macdonald et al., 2018). Crushed RMC samples also continued to release CH4 during their incubation, suggesting that wetting and mineral dissolution are important in this process (Fig. 6).
There are a few other mechanisms by which H2 might be produced, both of which involve surface silica radicals (Si⋅) generated during erosion of these samples. One mechanism involves H2 generation from the reaction of water with Si⋅ (Reactions R7 and R8; e.g. Hasegawa et al., 1995; Kita et al., 1982; Telling et al., 2015). The other mechanism is the potential reaction of hydroxyl functional groups (-OH) from within the crystal structure with Si⋅ (Reaction R18; Kameda et al., 2004). This mechanism generates H⋅, which react together to generate H2 (Reaction R8) from -OH-rich minerals and clays (Kameda et al., 2004) and was suggested as a potential mechanism for H2 generation during crushing of Robertson Glacier muddy carbonate samples (Macdonald et al., 2018). The gases measured during crushing and, in the case of RMC, during the 25 h incubation are likely a result of a combination of these mechanisms.
4.4 Erosion as a source of ammonium for nitrifiers
Nitrification involves the sequential oxidation of to (Reactions R19 and R20).
There is evidence of nitrification in several subglacial systems, including Svalbard (Hodson et al., 2010; Wynn et al., 2007), Robertson Glacier (Boyd et al., 2011) and Subglacial Lake Whillans (Christner et al., 2014). Foght et al. (2004) suggested that the source of to these environments is from in situ nitrogen fixation. However, incubations of subglacial sediments have been unsuccessful in detecting N fixation despite the detection of nitrogen-fixing bacteria (Boyd et al., 2011). Others have suggested exogenous sources of organic N to the system, such as wind-blown debris which was originally deposited on the glacier surface (Stibal et al., 2008). This debris is high in organic carbon and so is likely to contain significant concentrations of organic N (Boyd et al., 2011). It is then washed into the subglacial system through moulins and crevasses (Boyd et al., 2011; Stibal et al., 2008), where it could then be mineralised and utilised for nitrification. Saros et al. (2010) reported that, in catchments in the northern Rockies, lakes fed by snowpack and glacial meltwaters often contained 10 (and up to 200) times the amount of nitrate when compared to similar adjacent lakes fed only by snowpack meltwaters (SF lakes), implying that processes such as nitrification occurred in the glaciated or glacial catchment. Crushing of sedimentary rocks from alpine catchments and incubation with water at higher temperatures (20 ∘C) has shown the release of ammonium (G. G. Montross et al., 2013), similarly, from gneissic rocks from a glaciated catchment at 4 ∘C (Allen, 2019). Our results also indicate that rock comminution can act as a source of to the subglacial system. Crushing of glacial rock samples in the ball mill generates heat (Stone et al., 2022), which might have contributed to the measured. However, there was no correlation between and %N or %OC in the samples. Most likely, in solution is a direct result of the release of from clay minerals (Boyd, 1997) or from silicate mineral surfaces (Sugahara et al., 2017).
No matter the source, it is clear that abrasion increases the availability of for nitrification. Crude calculations can be made to estimate the potential generated from abrasion at a catchment level using suspended sediment fluxes. We estimate that Robertson Glacier (for RMC) and Longyearbreen (for SSv) could generate 3.5 and 4.7 , respectively (Supplement B). Incubations of subglacial samples from Robertson Glacier suggest that nitrifiers utilised 0.2 (Supplement B; Boyd et al., 2011) at peak activity, yet no nitrogen fixers were found. Therefore, subglacial abrasion could be a key source of to these ecosystems. This finding would be consistent with recent research that showed that bedrock N inputs from weathering are of a similar magnitude to atmospheric inputs in many terrestrial environments (Houlton et al., 2018). Further, H2O2 dissociates into O2 and H2O in the presence of catalase. Thus, H2O2 can be a source of dissolved oxygen which improves nitrification in systems with limited O2, such as wetlands (Dinakar et al., 2020), and could theoretically aid nitrification in subglacial systems.
A final consideration is how representative these experiments are of the production of crushed debris beneath glaciers and the potential for their reactivity during subsequent wetting. The mechanical crushing of rock in the laboratory is not analogous to subglacial comminution in many ways. First, the temperature is lower and the pressure is higher in the subglacial environment as a whole. The overall lower temperature may not be so much of an issue, since this is only a few degrees at most, but the temperatures that are reached during subglacial comminution, where rock fragments in ice may be dragged across the bed for example, are unknown but may be locally high at rock–rock contact points. Likewise, the temperatures reached in the laboratory crushing at rock–rock interaction points can be locally high and potentially much higher than in a subglacial environment where ice may thermally buffer against higher temperatures. These higher temperatures may increase the reaction rates of surface free radicals, increasing rates of H⋅ and H2 (Kita et al., 1982) and H2O2 (Stone et al., 2022) generation. The higher pressures of subglacial environments may not be of such great importance to the generation of free radicals, but they may influence the reaction pathways and products. Higher pressures favour interactions between gases which reduce the number of moles of gases in the system (e.g. 2 CO + O2 ⟶ 2 CO2: three moles of gas have been converted into two moles of gas).
Laboratory and in situ crushing beneath glaciers share at least one common element: minerals are sheared and so must have broken bonds on the surface. Comminution, and the deformation of pre-existing sediments, occurs often in the vicinity of meltwater beneath temperate-based and polythermal-based glaciers. Additionally, sediment entrainment onto cold-based glaciers may involve regelation waters or waters under the glacier freezing onto the base, trapping sediment as a consequence. Regelation is a common process at glacier beds, when pressure melting occurs as ice flows over protuberances or bumps in the bed. The ice melts at the high-pressure side of the bump, facing up-flow, and meltwater travels around the bump and refreezes on the lower-pressure down-flow side. Hence, overall, there is a reasonable probability that comminuted rock, otherwise called glacial flour, will come into contact with meltwater. However, the timescale in which this happens cannot be known with certainty, given the current understanding of glacial erosion. Timescales may be almost instantaneous in some areas of the bed, particularly if regelation and comminution occur at the same place and at the same time, but equally there may be much longer delays. This begs the question of how much water, and indeed whether even water vapour, is needed to initiate the surface-free-radical-catalysed reactions. Further, how comminuted minerals interact with ice during their entombment in debris-rich basal ice, for example, has yet to be examined. The simplicity of these experiments shows the potential of the comminution to supply chemicals for biogeochemical reactions in subglacial environments, but just how the experiments may map onto the production and wetting of subglacial sediment in real environments is more difficult to assess because pertinent subglacial parameters, some of which have been discussed here, are poorly known and quantified.
It is often difficult to disentangle the many confounding factors when trying to understand the interactions of competing processes in natural systems. Subglacial environments are no exception. We anticipated that the concentration of sulfide in sedimentary rocks would have a first-order impact on the amount of H2O2 that is generated when the rocks are crushed. Instead, we found that the presence or concentration of pyrite is not enough to predict the generation of H2O2 from abrasion. Our results suggest that the presence of carbonates and the pH of the solution will impact the concentrations of H2O2 generated. Other factors, such as SSA, will influence H2O2 generation at similar pH. This study suggests that erosion can provide sources of nutrients and energy (including CH4, H2, labile forms of OC as organic acids and ) to subglacial ecosystems. Further, our experiments highlight the importance of additional experiments which more closely represent conditions under glaciers and ice sheets (e.g. at pressure, wet crushing, several lower-energy crushing and wetting cycles) as a worthy pursuit. Caution should be taken to avoid over-extrapolation of these results, as these experiments use only a limited range of subglacial sedimentary rock and sediment samples. However, these results serve to highlight the potential role that abrasion plays in sustaining subglacial sedimentary-rock-hosted aquatic ecosystems.
All data presented in Figs. 1–6 are available online at EarthChem (https://doi.org/10.26022/IEDA/112762, Gill-Olivas et al., 2023).
The supplement related to this article is available online at: https://doi.org/10.5194/bg-20-929-2023-supplement.
BGO led the design of the study, assisted by MT and JT. Experimental work and laboratory analysis were performed by BGO. BGO prepared this paper with contributions from MT, JT and MS.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Moya MacDonald provided siltstone samples from Svalbard (SSv). We thank the SALSA Science Team for retrieval of the sediments from Mercer Subglacial Lake (SLM).
The research was supported by NERC (grant no. NE/S001670/1, CRUSH2LIFE; Beatriz Gill-Olivas, Martyn Tranter, Jon Telling) and NSF-OPP (grant no. 1543537; Mark Skidmore).
This paper was edited by Ji-Hyung Park and reviewed by Simon Bottrell and one anonymous referee.
Allen, J. J.: Glacial effects on stream water nitrate: an examination of paired catchments in southern Montana, Earth Sciences, Montana State University, 2019.
Anderson, S. P.: Glaciers show direct linkage between erosion rate and chemical weathering fluxes, Geomorphology, 67, 147–157, https://doi.org/10.1016/j.geomorph.2004.07.010, 2005.
Baga, A. N., Johnson, G. R. A., Nazhat, N. B., and Saadalla-Nazhat, R. A.: A simple spectrophotometric determination of hydrogen peroxide at low concentrations in aqueous solution, Anal. Chim. Acta, 204, 349–353, https://doi.org/10.1016/S0003-2670(00)86374-6, 1988.
Bak, E. N., Zafirov, K., Merrison, J. P., Jensen, S. J. K., Nornberg, P., Gunnlaugsson, H. P., and Finster, K.: Production of reactive oxygen species from abraded silicates. Implications for the reactivity of the Martian soil, Earth Planet. Sc. Lett., 473, 113–121, https://doi.org/10.1016/j.epsl.2017.06.008, 2017.
Bataineh, H., Pestovsky, O., and Bakac, A.: pH-induced mechanistic changeover from hydroxyl radicals to iron(iv) in the Fenton reaction, Chem. Sci., 3, 1594–1599, https://doi.org/10.1039/C2SC20099F, 2012.
Borda, M. J., Elsetinow, A. R., Schoonen, M. A., and Strongin, D. R.: Pyrite-Induced Hydrogen Peroxide Formation as a Driving Force in the Evolution of Photosynthetic Organisms on an Early Earth, Astrobiology, 1, 283–288, https://doi.org/10.1089/15311070152757474, 2001.
Borda, M. J., Elsetinow, A. R., Strongin, D. R., and Schoonen, M. A.: A mechanism for the production of hydroxyl radical at surface defect sites on pyrite, Geochim. Cosmochim. Ac., 67, 935–939, https://doi.org/10.1016/S0016-7037(02)01222-X, 2003.
Boyd, E. S., Skidmore, M., Mitchell, A. C., Bakermans, C., and Peters, J. W.: Methanogenesis in subglacial sediments, Env. Microbiol. Rep., 2, 685–692, https://doi.org/10.1111/j.1758-2229.2010.00162.x, 2010.
Boyd, E. S., Lange, R. K., Mitchell, A. C., Havig, J. R., Hamilton, T. L., Lafrenière, M. J., Shock, E. L., Peters, J. W., and Skidmore, M.: Diversity, Abundance, and Potential Activity of Nitrifying and Nitrate-Reducing Microbial Assemblages in a Subglacial Ecosystem, Appl. Environ. Microb., 77, 4778–4787, https://doi.org/10.1128/AEM.00376-11, 2011.
Boyd, S. R.: Determination of the ammonium content of potassic rocks and minerals by capacitance manometry: a prelude to the calibration of FTIR microscopes, Chem. Geol., 137, 57–66, https://doi.org/10.1016/S0009-2541(96)00150-7, 1997.
Campbell, T., Patterson, M. O., Skidmore, M. L., Leventer, A., Michaud, A. B., Rosenheim, B. E., Harwood, D. M., Dore, J. E., Tranter, M., Venturelli, R., and Priscu, J. C.: Physical and chemical characterization of sediments from Mercer Subglacial Lake, West Antarctica, AGU Fall Meeting, Moscone Centre, San Francisco CA (USA), 9–13 December 2019, C53B-1342, 2019.
Carter, S. P., Fricker, H. A., and Siegfried, M. R.: Evidence of rapid subglacial water piracy under Whillans Ice Stream, West Antarctica, J. Glaciol., 59, 1147–1162, https://doi.org/10.3189/2013JoG13J085, 2013.
Christner, B. C., Priscu, J. C., Achberger, A. M., Barbante, C., Carter, S. P., Christianson, K., Michaud, A. B., Mikucki, J. A., Mitchell, A. C., Skidmore, M. L., Vick-Majors, T. J., and WISSARD Science Team: A microbial ecosystem beneath the West Antarctic ice sheet, Nature, 512, 310–313, https://doi.org/10.1038/nature13667, 2014.
Dinakar, M., Tao, W., and Daley, D.: Using hydrogen peroxide to supplement oxygen for nitrogen removal in constructed wetlands, Journal of Environmental Chemical Engineering, 8, 104517, https://doi.org/10.1016/j.jece.2020.104517, 2020.
Dunham, E. C., Dore, J. E., Skidmore, M. L., Roden, E. E., and Boyd, E. S.: Lithogenic hydrogen supports microbial primary production in subglacial and proglacial environments, P. Natl. Acad. Sci. USA, 118, e2007051117, https://doi.org/10.1073/pnas.2007051117, 2021.
Edgar, J. O., Gilmour, K., White, M. L., Abbott, G. D., and Telling, J.: Aeolian driven oxidant and hydrogen generation in Martian regolith: The role of mineralogy and abrasion temperature, Earth Planet. Sc. Lett., 579, 117361, https://doi.org/10.1016/j.epsl.2021.117361, 2022.
Elvevold, S., Dallmann, W., and Blomeier, D.: Geology of Svalbard, Norwegian Polar Institute/Norsk Polarinstitutt, Tromsø, Norway, 2007.
Foght, J., Aislabie, J., Turner, S., Brown, C. E., Ryburn, J., Saul, D. J., and Lawson, W.: Culturable bacteria in subglacial sediments and ice from two Southern Hemisphere glaciers, Microb. Ecol., 47, 329–340, https://doi.org/10.1007/s00248-003-1036-5, 2004.
Folk, R. L. and Ward, W. C.: Brazos River bar [Texas]; a study in the significance of grain size parameters, J. Sediment. Res., 27, 3–26, https://doi.org/10.1306/74d70646-2b21-11d7-8648000102c1865d, 1957.
Gil-Lozano, C., Losa-Adams, E., Davila, A. F., and Gago-Duport, L.: Pyrite nanoparticles as a Fenton-like reagent for in situ remediation of organic pollutants, Beilstein J. Nanotech., 5, 855–864, https://doi.org/10.3762/bjnano.5.97, 2014.
Gil-Lozano, C., Davila, A. F., Losa-Adams, E., Fairén, A. G., and Gago-Duport, L.: Quantifying Fenton reaction pathways driven by self-generated H2O2 on pyrite surfaces, Sci. Rep.-UK, 7, 43703, https://doi.org/10.1038/srep43703, 2017.
Gill-Olivas, B., Telling, J., Tranter, M., Skidmore, M., Christner, B., O'Doherty, S., and Priscu, J.: Subglacial erosion has the potential to sustain microbial processes in Subglacial Lake Whillans, Antarctica, Communications Earth & Environment, 2, 134, https://doi.org/10.1038/s43247-021-00202-x, 2021.
Gill-Olivas, B., Telling, J., and Tranter, M.: Hydrogen peroxide, solute and methane released from crushing sedimentary rocks, Version 1.0, Interdisciplinary Earth Data Alliance (IEDA) [data set], https://doi.org/10.26022/IEDA/112762, 2023.
Harris, D., Horwáth, W. R., and van Kessel, C.: Acid fumigation of soils to remove carbonates prior to total organic carbon or Carbon-13 isotopic analysis, Soil Sci. Soc. Am. J., 65, 1853–1856, https://doi.org/10.2136/sssaj2001.1853, 2001.
Hasegawa, M., Ogata, T., and Sato, M.: Mechano-radicals produced from ground quartz and quartz glass, Powder Technol., 85, 269–274, https://doi.org/10.1016/0032-5910(96)80150-1, 1995.
He, H., Wu, X., Xian, H., Zhu, J., Yang, Y., Lv, Y., Li, Y., and Konhauser, K. O.: An abiotic source of Archean hydrogen peroxide and oxygen that pre-dates oxygenic photosynthesis, Nat. Commun., 12, 6611, https://doi.org/10.1038/s41467-021-26916-2, 2021.
Hodson, A., Tranter, M., and Vatne, G.: Contemporary rates of chemical denudation and atmospheric CO2 sequestration in glacier basins: An Arctic perspective, Earth Surf. Proc. Land., 25, 1447–1471, https://doi.org/10.1002/1096-9837(200012)25:13<1447::aid-esp156>3.0.co;2-9, 2000.
Hodson, A., Anesio, A. M., Tranter, M., Fountain, A., Osborn, M., Priscu, J., Laybourn-Parry, J., and Sattler, B.: Glacial ecosystems, Ecol. Monogr., 78, 41–67, https://doi.org/10.1890/07-0187.1, 2008.
Hodson, A., Roberts, T. J., Engvall, A.-C., Holmén, K., and Mumford, P.: Glacier ecosystem response to episodic nitrogen enrichment in Svalbard, European High Arctic, Biogeochemistry, 98, 171–184, https://doi.org/10.1007/s10533-009-9384-y, 2010.
Hodson, T. O., Powell, R. D., Brachfeld, S. A., Tulaczyk, S., Scherer, R. P., and WISSARD Science Team: Physical processes in Subglacial Lake Whillans, West Antarctica: Inferences from sediment cores, Earth Planet. Sc. Lett., 444, 56–63, https://doi.org/10.1016/j.epsl.2016.03.036, 2016.
Hood, E., Fellman, J., Spencer, R. G. M., Hernes, P. J., Edwards, R., D'Amore, D., and Scott, D.: Glaciers as a source of ancient and labile organic matter to the marine environment, Nature, 462, 1044–1047, https://doi.org/10.1038/nature08580, 2009.
Houlton, B. Z., Morford, S. L., and Dahlgren, R. A.: Convergent evidence for widespread rock nitrogen sources in Earth's surface environment, Science, 360, 58–62, https://doi.org/10.1126/science.aan4399, 2018.
Illes, E., Mizrahi, A., Marks, V., and Meyerstein, D.: Carbonate-radical-anions, and not hydroxyl radicals, are the products of the Fenton reaction in neutral solutions containing bicarbonate, Free Radical Bio. Med., 131, 1–6, https://doi.org/10.1016/j.freeradbiomed.2018.11.015, 2019.
Kameda, J., Saruwatari, K., and Tanaka, H.: H2 generation during dry grinding of kaolinite, J. Colloid Interf. Sci., 275, 225–228, https://doi.org/10.1016/j.jcis.2004.02.014, 2004.
Kaštovská, K., Stibal, M., Šabacká, M., Černá, B., Šantrůčková, H., and Elster, J.: Microbial community structure and ecology of subglacial sediments in two polythermal Svalbard glaciers characterized by epifluorescence microscopy and PLFA, Polar Biol., 30, 277–287, https://doi.org/10.1007/s00300-006-0181-y, 2007.
Kita, I., Matsuo, S., and Wakita, H.: H2 generation by reaction between H2O and crushed rock – An experimental-study on H2 degassing from the active fault zone, J. Geophys. Res., 87, 789–795, https://doi.org/10.1029/JB087iB13p10789, 1982.
Kohler, T. J., Žárský, J. D., Yde, J. C., Lamarche-Gagnon, G., Hawkings, J. R., Tedstone, A. J., Wadham, J. L., Box, J. E., Beaton, A. D., and Stibal, M.: Carbon dating reveals a seasonal progression in the source of particulate organic carbon exported from the Greenland Ice Sheet, Chem. Geol., 44, 6209–6217, https://doi.org/10.1002/2017GL073219, 2017.
Lanoil, B., Skidmore, M., Priscu, J. C., Han, S., Foo, W., Vogel, S. W., Tulaczyk, S., and Engelhardt, H.: Bacteria beneath the West Antarctic Ice Sheet, Environ. Microbiol., 11, 609–615, https://doi.org/10.1111/j.1462-2920.2008.01831.x, 2009.
Lawson, E. C., Wadham, J. L., Tranter, M., Stibal, M., Lis, G. P., Butler, C. E. H., Laybourn-Parry, J., Nienow, P., Chandler, D., and Dewsbury, P.: Greenland Ice Sheet exports labile organic carbon to the Arctic oceans, Biogeosciences, 11, 4015–4028, https://doi.org/10.5194/bg-11-4015-2014, 2014.
Le, P. T. T. and Boyd, C. E.: Comparison of Phenate and Salicylate Methods for Determination of Total Ammonia Nitrogen in Freshwater and Saline Water, J. World Aquacult. Soc., 43, 885–889, https://doi.org/10.1111/j.1749-7345.2012.00616.x, 2012.
Lerman, A.: Dissolution of Feldspars, in: Geochemical Processes Water and Sediment Environments, Robert E. Krieager Publishing Company, Malabar, Florida, 244–248, ISBN: 9780471032632, 1988.
Macdonald, M. L., Wadham, J. L., Telling, J., and Skidmore, M. L.: Glacial Erosion Liberates Lithologic Energy Sources for Microbes and Acidity for Chemical Weathering Beneath Glaciers and Ice Sheets, Front. Earth Sci., 6, 15, https://doi.org/10.3389/feart.2018.00212, 2018.
Madigan, M. T., Bender, K. S., Buckley, D. H., Sattley, W. M., and Stahl, D. A.: Brock Biology of Microorganisms, 16th edn., Pearson, Harlow, United Kingdom, ISBN: 9781292404790, 2020.
McMechan, M. E.: Geology, Peter Lougheed Provincial Park, west of fifth meridian, Alberta, “A” Series Map 1920A, Geological Survey of Canada, https://doi.org/10.4095/209966, 1998.
Medina-Cordoba, L. K., Valencia-Mosquera, L. L., Tarazona-Diaz, G. P., and Arias-Palacios, J. D. C.: Evaluation of the efficacy of a hydrogen peroxide disinfectant, International Journal of Pharmacy and Pharmaceutical Sciences, 10, 104–108, https://doi.org/10.22159/ijpps.2018v10i10.24652, 2018.
Michaud, A. B., Skidmore, M. L., Mitchell, A. C., Vick-Majors, T. J., Barbante, C., Turetta, C., vanGelder, W., and Priscu, J. C.: Solute sources and geochemical processes in Subglacial Lake Whillans, West Antarctica, Geology, 44, 347–350, https://doi.org/10.1130/g37639.1, 2016.
Michaud, A. B., Dore, J. E., Achberger, A. M., Christner, B. C., Mitchell, A. C., Skidmore, M. L., Vick-Majors, T. J., and Priscu, J. C.: Microbial oxidation as a methane sink beneath the West Antarctic Ice Sheet, Nat. Geosci., 10, 582–586, https://doi.org/10.1038/ngeo2992, 2017.
Montross, G. G., McGlynn, B. L., Montross, S. N., and Gardner, K. K.: Nitrogen production from geochemical weathering of rocks in southwest Montana, USA, J. Geophys. Res.-Biogeo., 118, 1068–1078, https://doi.org/10.1002/jgrg.20085, 2013.
Montross, S. N., Skidmore, M., Tranter, M., Kivimäki, A.-L., and Parkes, R. J.: A microbial driver of chemical weathering in glaciated systems, Geology, 41, 215–218, https://doi.org/10.1130/G33572.1, 2013.
Moore, D. S., Notz, W. I., and Flinger, M. A.: The basic practice of statistics, 6th edn., W. H. Freeman and Company, New York, NY, ISBN: 1464117667, 2013.
Nesbitt, H. W., Bancroft, G. M., Pratt, A. R., and Scaini, M. J.: Sulfur and iron surface states on fractured pyrite surfaces, Am. Mineral., 83, 1067–1076, https://doi.org/10.2138/am-1998-9-1015, 1998.
Plummer, L. N., Wigley, T. M., and Parkhurst, D. L.: The kinetics of calcite dissolution in CO2-water systems at 5 degrees to 60 ∘C and 0.0 to 1.0 atm CO2, Am. J. Chem., 278, 179–216, 1978.
Priscu, J. C., Tulaczyk, S., Studinger, M., Kennicutt II, M. C., Christner, B., and Foreman, C.: Antarctic subglacial water: origin, evolution, and ecology, in: Polar Lakes and Rivers, edited by: Vincent, W. F. and Laybourn-Parry, J., Oxford University Press, Oxford, UK, 119–136, 9780199213887, 2008.
Priscu, J. C., Kalin, J., Winans, J., Campbell, T., Siegfried, M. R., Skidmore, M., Dore, J. E., Leventer, A., Harwood, D. M., Duling, D., Zook, R., Burnett, J., Gibson, D., Krula, E., Mironov, A., McManis, J., Roberts, G., Rosenheim, B. E., Christner, B. C., Kasic, K., Fricker, H. A., Lyons, W. B., Barker, J., Bowling, M., Collins, B., Davis, C., Gagnon, A., Gardner, C., Gustafson, C., Kim, O.-S., Li, W., Michaud, A., Patterson, M. O., Tranter, M., Venturelli, R., Vick-Majors, T., and Elsworth, C.: Scientific access into Mercer Subglacial Lake: scientific objectives, drilling operations and initial observations, Ann. Glaciol., 62, 340–352, https://doi.org/10.1017/aog.2021.10, 2021.
Rimstidt, J. D. and Vaughan, D. J.: Pyrite oxidation: a state-of-the-art assessment of the reaction mechanism, Geochim. Cosmochim. Ac., 67, 873–880, https://doi.org/10.1016/S0016-7037(02)01165-1, 2003.
Saros, J. E., Rose, K. C., Clow, D. W., Stephens, V. C., Nurse, A. B., Arnett, H. A., Stone, J. R., Williamson, C. E., and Wolfe, A. P.: Melting Alpine Glaciers Enrich High-Elevation Lakes with Reactive Nitrogen, Environ. Sci. Technol., 44, 4891–4896, https://doi.org/10.1021/es100147j, 2010.
Saruwatari, K., Kameda, J., and Tanaka, H.: Generation of hydrogen ions and hydrogen gas in quartz-water crushing experiments: an example of chemical processes in active faults, Phys. Chem. Miner., 31, 176–182, https://doi.org/10.1007/s00269-004-0382-2, 2004.
Skidmore, M.: Microbial communities in Antarctic subglacial aquatic environments, American Geophysical Union Geophysical Monograph Series, American Geophysical Union, Washington, DC, 192, 61–81, 2011.
Skidmore, M., Tranter, M., Tulaczyk, S., and Lanoil, B.: Hydrochemistry of ice stream beds – evaporitic or microbial effects?, Hydrol. Process., 24, 517–523, https://doi.org/10.1002/hyp.7580, 2010.
Souchez, R., Jouzel, J., Landais, A., Chappellaz, J., Lorrain, R., and Tison, J.-L.: Gas isotopes in ice reveal a vegetated central Greenland during ice sheet invasion, Geophys. Res. Lett., 33, L24503, https://doi.org/10.1029/2006GL028424, 2006.
Stibal, M., Tranter, M., Benning, L. G., and Řehák, J.: Microbial primary production on an Arctic glacier is insignificant in comparison with allochthonous organic carbon input, Environ. Microbiol., 10, 2172–2178, https://doi.org/10.1111/j.1462-2920.2008.01620.x, 2008.
Stibal, M., Wadham, J. L., Lis, G. P., Telling, J., Pancost, R. D., Dubnick, A., Sharp, M. J., Lawson, E. C., Butler, C. E. H., Hasan, F., Tranter, M., and Anesio, A. M.: Methanogenic potential of Arctic and Antarctic subglacial environments with contrasting organic carbon sources, Glob. Change Biol., 18, 3332–3345, https://doi.org/10.1111/j.1365-2486.2012.02763.x, 2012.
Stillings, M., Lunn, R. J., Pytharouli, S., Shipton, Z. K., Kinali, M., Lord, R., and Thompson, S.: Microseismic Events Cause Significant pH Drops in Groundwater, Geophys. Res. Lett., 48, e2020GL089885, https://doi.org/10.1029/2020GL089885, 2021.
Stone, J., Edgar, J. O., Gould, J. A., and Telling, J.: Tectonically-driven oxidant production in the hot biosphere, Nat. Commun., 13, 4529, https://doi.org/10.1038/s41467-022-32129-y, 2022.
Sugahara, H., Takano, Y., Ogawa, N. O., Chikaraishi, Y., and Ohkouchi, N.: Nitrogen Isotopic Fractionation in Ammonia during Adsorption on Silicate Surfaces, ACS Earth and Space Chemistry, 1, 24–29, https://doi.org/10.1021/acsearthspacechem.6b00006, 2017.
Suzuki, N., Saito, H., and Hoshino, T.: Hydrogen gas of organic origin in shales and metapelites, Int. J. Coal Geol., 173, 227–236, https://doi.org/10.1016/j.coal.2017.02.014, 2017.
Takehiro, H., Shinsuke, K., and Katsuhiko, S.: Mechanoradical H2 generation during simulated faulting: Implications for an earthquake-driven subsurface biosphere, Geophys. Res. Lett., 38, L17303, https://doi.org/10.1029/2011GL048850, 2011.
Telling, J., Boyd, E. S., Bone, N., Jones, E. L., Tranter, M., MacFarlane, J. W., Martin, P. G., Wadham, J. L., Lamarche-Gagnon, G., Skidmore, M. L., Hamilton, T. L., Hill, E., Jackson, M., and Hodgson, D. A.: Rock comminution as a source of hydrogen for subglacial ecosystems, Nat. Geosci., 8, 851–855, https://doi.org/10.1038/ngeo2533, 2015.
Tranter, M.: Geochemical Weathering in Glacial and Proglacial Environments, in: Treatise on Geochemistry, edited by: Holland, H. D. and Turekian, K. K., Elsevier-Permagon, Oxford, 189–205, https://doi.org/10.1016/B0-08-043751-6/05078-7, 2003.
Tranter, M.: Biogeochemistry Microbes eat rock under ice, Nature, 512, 256–257, https://doi.org/10.1038/512256a, 2014.
Tranter, M.: Grand challenge for low temperature and pressure geochemistry – sparks in the dark, on Earth, Mars, and throughout the Galaxy, Earth Sci., 3, 69, https://doi.org/10.3389/feart.2015.00069, 2015.
Tranter, M., Brown, G., Raiswell, R., Sharp, M., and Gurnell, A.: A conceptual model of solute acquisition by Alpine glacial meltwaters, J. Glaciol., 39, 573–581, https://doi.org/10.3189/S0022143000016464, 1993.
Tranter, M., Sharp, M. J., Lamb, H. R., Brown, G. H., Hubbard, B. P., and Willis, I. C.: Geochemical weathering at the bed of Haut Glacier d'Arolla, Switzerland – a new model, Hydrol. Process., 16, 959–993, https://doi.org/10.1002/hyp.309, 2002a.
Tranter, M., Huybrechts, P., Munhoven, G., Sharp, M. J., Brown, G. H., Jones, I. W., Hodson, A. J., Hodgkins, R., and Wadham, J. L.: Direct effect of ice sheets on terrestrial bicarbonate, sulphate and base cation fluxes during the last glacial cycle: minimal impact on atmospheric CO2 concentrations, Chem. Geol., 190, 33–44, https://doi.org/10.1016/s0009-2541(02)00109-2, 2002b.
Tranter, M., Skidmore, M., and Wadham, J.: Hydrological controls on microbial communities in subglacial environments, Hydrol. Process., 19, 995–998, https://doi.org/10.1002/hyp.5854, 2005.
Tulaczyk, S., Kamb, B., Scherer, R. P., and Engelhardt, H. F.: Sedimentary processes at the base of a West Antarctic ice stream: Constraints from textural and compositional properties of subglacial debris, J. Sediment. Res., 68, 487–496, 1998.
Viollier, E., Inglett, P. W., Hunter, K., Roychoudhury, A. N., and Van Cappellen, P.: The ferrozine method revisited: Fe(II)/Fe(III) determination in natural waters, Appl. Geochem., 15, 785–790, https://doi.org/10.1016/s0883-2927(99)00097-9, 2000.
Wadham, J. L., Tranter, M., Skidmore, M., Hodson, A. J., Priscu, J., Lyons, W. B., Sharp, M., Wynn, P., and Jackson, M.: Biogeochemical weathering under ice: Size matters, Global Biogeochem. Cy., 24, 11, https://doi.org/10.1029/2009gb003688, 2010.
Wadham, J. L., Hawkings, J. R., Tarasov, L., Gregoire, L. J., Spencer, R. G. M., Gutjahr, M., Ridgwell, A., and Kohfeld, K. E.: Ice sheets matter for the global carbon cycle, Nat. Commun., 10, 3567, https://doi.org/10.1038/s41467-019-11394-4, 2019.
Wakita, H., Nakamura, Y., Kita, I., Fujii, N., and Notsu, K.: Hydrogen release – New indicator of fault activity, Science, 210, 188–190, https://doi.org/10.1126/science.210.4466.188, 1980.
Wiebe, R. and Gaddy, V. L.: The Solubility of Hydrogen in Water at 0, 50, 75 and 100∘ from 25 to 1000 Atmospheres, J. Am. Chem. Soc., 56, 76–79, https://doi.org/10.1021/ja01316a022, 1934.
Wynn, P. M., Hodson, A. J., Heaton, T. H. E., and Chenery, S. R.: Nitrate production beneath a High Arctic glacier, Svalbard, Chem. Geol., 244, 88–102, https://doi.org/10.1016/j.chemgeo.2007.06.008, 2007.
Zhou, S., Zhang, D., Wang, H., and Li, X.: A modified BET equation to investigate supercritical methane adsorption mechanisms in shale, Mar. Petrol. Geol., 105, 284–292, https://doi.org/10.1016/j.marpetgeo.2019.04.036, 2019.