the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Apr 2021
| 06 Apr 2021
Effects of tidal influence on the structure and function of prokaryotic communities in the sediments of a pristine Brazilian mangrove
Carolina Oliveira de Santana
Pieter Spealman
Vânia Maria Maciel Melo
David Gresham
Taíse Bomfim de Jesus
Fabio Alexandre Chinalia
Mangrove forests are ecosystems that constitute a large portion of the world's coastline and span tidal zones below, between, and above the waterline, and the ecosystem as a whole is defined by the health of these tidal microhabitats. However, we are only beginning to understand tidal-zone microbial biodiversity and the role of these microbiomes in nutrient cycling. While extensive research has characterized microbiomes in pristine vs. anthropogenically impacted mangroves, these have, largely, overlooked differences in tidal microhabitats (sublittoral, intertidal, and supralittoral). Unfortunately, the small number of studies that have sought to characterize mangrove tidal zones have occurred in impacted biomes, making interpretation of the results difficult. Here, we characterized prokaryotic populations and their involvement in nutrient cycling across the tidal zones of a pristine mangrove within a Brazilian Environmental Protection Area of the Atlantic Forest. We hypothesized that the tidal zones in pristine mangroves are distinct microhabitats, which we defined as distinct regions that present spatial variations in the water regime and other environmental factors, and as such, these are composed of different prokaryotic communities with distinct functional profiles. Samples were collected in triplicate from zones below, between, and above the tidal waterline. Using 16S ribosomal RNA (rRNA) gene amplicon sequencing, we found distinct prokaryotic communities with significantly diverse nutrient-cycling functions, as well as specific taxa with varying contributions to functional abundances between zones. Where previous research from anthropogenically impacted mangroves found the intertidal zone to have high prokaryotic diversity and be functionally enriched in nitrogen cycling, we find that the intertidal zone from pristine mangroves has the lowest diversity and no functional enrichment, relative to the other tidal zones. The main bacterial phyla in all samples were Firmicutes, Proteobacteria, and Chloroflexi while the main archaeal phyla were Crenarchaeota and Thaumarchaeota. Our results differ slightly from other studies where Proteobacteria is the main phyla in mangrove sediments and Firmicutes makes up only a small percentage of the communities. Salinity and organic matter were the most relevant environmental factors influencing these communities. Bacillaceae was the most abundant family at each tidal zone and showed potential to drive a large proportion of the cycling of carbon, nitrogen, phosphorus, and sulfur. Our findings suggest that some aspects of mangrove tidal zonation may be compromised by human activity, especially in the intertidal zone.
- Article
(3779 KB) - Full-text XML
-
Supplement
(7971 KB) - BibTeX
- EndNote





Mangrove ecosystems exist at the interface of land and sea and constitute a large portion of tropical and subtropical coastlines, spanning 118 countries and approximately 137 000 km2 (Giri et al., 2011). Within these ecosystems, the recycling of elements occurs via tightly coupled exchanges between plants, microorganisms, and microbes, with Bacteria and Archaea playing important roles in the biogeochemical cycling of carbon, nitrogen, and phosphate (Imchen et al., 2017; Lin et al., 2019; Alongi, 1988; Reis et al., 2017; Holguin et al., 2001). Mangroves span tidal zones that are characterized by periodic tidal flooding, such that environmental conditions such as salinity vary greatly across small spatiotemporal scales. We refer to the differences in microhabitats and biodiversity in mangrove sediments as zonation, and we consider the mangroves to be divided into three main tidal zones, where the sublittoral zone is always immersed, while the intertidal zone is exposed to daily variations in water content and the supralittoral zone is the region that presents mangrove vegetation but normally is above the sea level. With each tidal cycle the levels of nutrients, oxygen, and salinity fluctuate, resulting in frequent anaerobic conditions and a wide range of redox potentials in the sediments (Holguin et al., 2001; Andreote et al., 2012; Marcos et al., 2018; Lin et al., 2019). While there has been little research on microbial diversity within mangrove tidal zones, research in non-mangrove tidal ecosystems has found distinct microbiomes with significantly different biogeochemistries (Musat et al., 2006; Boehm et al., 2014; Azzoni et al., 2015). Understanding the interrelationship of mangrove tidal zones, their microbial populations, and their role in nutrient cycling is a key component in understanding mangrove adaptation and conservation (Coldren et al., 2019; Saintilan et al., 2020; Shiau and Chiu, 2020; Allard et al., 2020).
Numerous studies have sought to characterize mangrove microbiomes in both pristine and anthropogenically impacted areas (see Supplement file 2). This has led to an increased understanding of the sensitivity of mangrove microbiomes to perturbations, such as pollution (Peixoto et al., 2011; Andreote et al., 2012; Nogueira et al., 2015), environmental degradation (Marcial Gomes et al., 2008; Tong et al., 2019; Cabral et al., 2018), and rising sea levels (Ceccon et al., 2019; Yun et al., 2017; Saintilan et al., 2020). Recently, additional work has sought to understand the diversity of microbial populations across microhabitats defined by distinct biotic and abiotic characteristics (Luis et al., 2019; Lv et al., 2016; Zhang et al., 2018; Rocha et al., 2016). While the particular effects of these perturbations vary from site to site, broadly these result in significantly altered population structures with substantially different functional profiles. This is important to consider since research in mangrove tidal-zone microhabitats (Zhang et al., 2018; Jiang et al., 2013; Zhou et al., 2017) has so far been carried out in anthropogenically impacted sites, thus confounding the interpretation of these findings.
In Brazil, mangrove forests are primarily composed of the genera Rhizophora, Avicennia, Laguncularia, and Conocarpus (Pupin and Nahas, 2014) and are part of the Atlantic Forest, one of the most biodiverse biomes on the planet. Unfortunately, the Atlantic Forest and associated ecosystems are highly threatened by anthropogenic disturbances, resulting in a severe decline from the forest's approximate pre-colonial area (Brooks et al., 1999; Ditt et al., 2013). However, in the southern part of Bahia State, Brazil, a significant fragment of the Atlantic Forest remains preserved within the Environmental Protection Area of Pratigi (MMA, Ministério do Meio Ambiente, 2004). Recent studies on the area have shown that preservation efforts have been generally effective, resulting in a high environmental quality relative to most mangrove forests, both in Brazil and globally (Ditt et al., 2013; Lopes, 2011; Mascarenhas et al., 2019). This preserved area constitutes an increasingly rare site for the understanding of the ecology of unimpacted mangrove forests.
In this study we aimed to characterize the prokaryotic microbiota from sediments of three tidal zones in a pristine mangrove located in the Serinhaém estuary, within the Environmental Protection Area of Pratigi, using 16S ribosomal RNA (rRNA) gene amplicon sequencing. Considering that mangrove zonation is driven, primarily, by tidal variation, we hypothesized that sediments of different tidal zones would differ significantly in the diversity, composition, and functioning of prokaryotic communities as a result of differences in the physicochemical properties affected by tides. We also hypothesized that the intertidal zone, being a highly dynamic environment, would have the highest diversity as reported previously (Zhang et al., 2018; Brose and Hillebrand, 2016). Several important ecological consequences follow on from high levels of biodiversity, such as increased functional redundancy and increased resilience to environmental perturbations (Girvan et al., 2005). However, unlike the findings from impacted mangroves (Zhang et al., 2018; Jiang et al., 2013) we found the intertidal zone to be the least diverse, suggesting that it may, in fact, be more sensitive to anthropogenic disruption. Our study provides insight into the role of microbes in the functioning of mangrove forests and establishes a baseline for monitoring the health of this important ecosystem, since this information is still scarce for pristine mangrove sites.
This work was conducted before a massive oil spill occurred off the coastline of Brazil in August 2019, impacting hundreds of miles of coastline including the Serinhaém estuary, where this research was conducted. This work therefore serves as a baseline measure of the prokaryotic communities of the tidal zones of what was a pristine mangrove forest. We hope that this will spur subsequent research into the effects that anthropogenic interferences have on mangrove ecosystems.
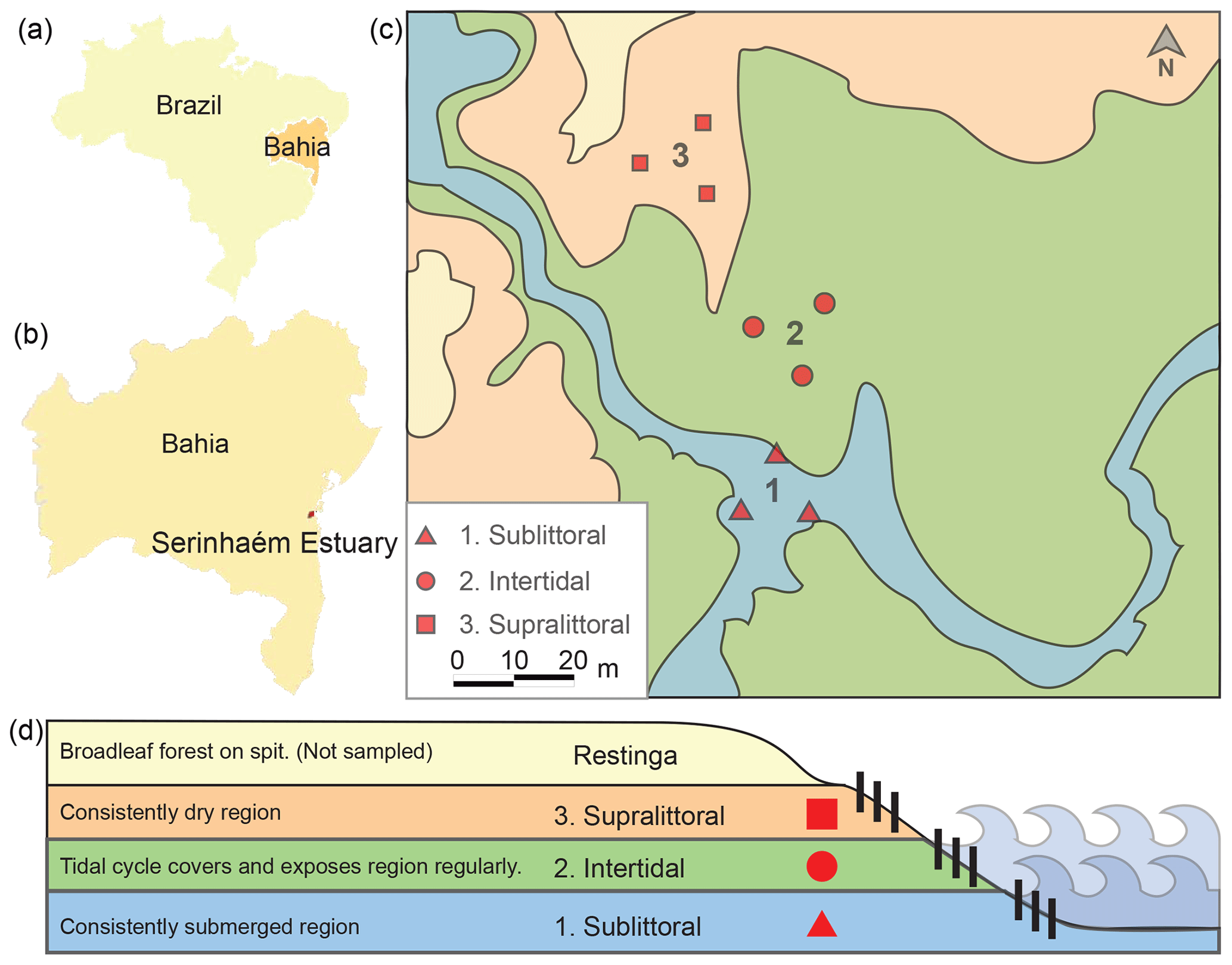
Figure 1Map and schematic of sediment sampling sites. Here we show the locations of the sampling sites relative to Brazil (a) and Bahia (b). A picture shows the relation of the three sampling sites within each zone (1 sublittoral, 2 intertidal, 3 supralittoral; c). A schematic shows the topographic and tidal relation of each sampling site (d).
2.1 Study area
The Serinhaém estuary is located in the Low South Region of Bahia State, Brazil (Fig. 1), between the coordinates 13∘35′ and 14∘10′ S, 39∘40′ and 38∘50′ W. The estuary is within the Environmental Protection Area of Pratigi, an Atlantic Forest region with a total area of 85 686 ha, enclosing a 32 km long portion of the Juliana River and emptying directly into Camamu Bay (Corrêa-Gomes et al., 2005).
2.2 Sampling and DNA extraction
For clarity, here we refer to the location of a sample as a “site” and the collection of sites within a tidal zone as a “zone”. Samples were collected from three tidal zones (centered around 13∘42′59.0′′ S, 39∘01′35.9′′ W) in the Serinhaém estuary in July 2018 during the morning low-tide period. No sites exhibited signs of anthropogenic disturbance or pollution. The three collection zones were chosen based on tidal influence: sublittoral, intertidal, and supralittoral regions (Fig. 1). We evaluated tidal zones based on water level at morning low tide; additional factors were also considered to differentiate between the intertidal and supralittoral zones, such as the presence of surface soil moisture, proximity to restinga vegetation, and the direction of a local guide. From each tidal zone, three samples of superficial sediments (top 10 cm of the surface layer) were collected with a cylindrical sediment core sampler. Precautions were taken to avoid the disruption of rhizospheres associated with vegetation, which could otherwise lead to contamination of the soil by rhizosphere-specific microbes. Sample sites were located at a minimum of 15 m from each other in a triangle. Plant and other organic material was subsequently removed from core samples.
Physical–chemical parameters were measured using a multiparameter monitoring system (YSI model 85, Columbus). Metal concentration analysis had been previously performed by our laboratory (da Silva Pereira, 2016) and found no significant difference in metal concentrations relative to background within the Serinhaém estuary. For each sample an aliquot was taken for DNA extraction, while the remainder was used for measuring organic matter content using “loss on ignition” (Nelson and Sommers, 1996). The total genomic DNA was extracted from 0.25 g of sediment using the PowerSoil DNA Isolation Kit (Qiagen, Carlsbad, CA, USA).
2.3 Library preparation and sequencing
After DNA extraction, we amplified the V4 region of the prokaryotic 16S rRNA gene using PCR with the primer pair 515F-Y (Parada et al., 2016) and 806R-XT (Caporaso et al., 2011) using KAPA HiFi for 25 cycles (see Supplement methods, file 1). Illumina sequencing adapters and dual-index barcodes were added to the amplicon target using Nextera XT according to the manufacturer's directions (Illumina, San Diego, CA, USA). DNA sequencing was performed using the Illumina MiSeq platform, V2 kit (300 cycles).
2.4 Data analysis
2.4.1 Sequence trimming, denoising, and amplicon sequence variant (ASV) clustering
Trimmomatic (Bolger et al., 2014) was used to filter and trim demultiplexed sequences. Paired reads were combined using QIIME 2 for reads with an overlap of greater than 9, while paired reads with an overlap of greater than 6 were combined using a custom script (see Supplement methods, file 1). Reads were denoised using DADA2 (Callahan et al., 2016) in QIIME 2 (Bolyen et al., 2019) and then clustered into amplicon sequence variants (ASVs). Alpha rarefaction was calculated using QIIME 2 (Fig. S1 in the Supplement). We performed a variety of alpha-diversity (Figs. S2–S4 in the Supplement), beta-diversity (Fig. S5 in the Supplement), and hierarchical correlation clustering (Fig. S6 in the Supplement) tests using QIIME 2.
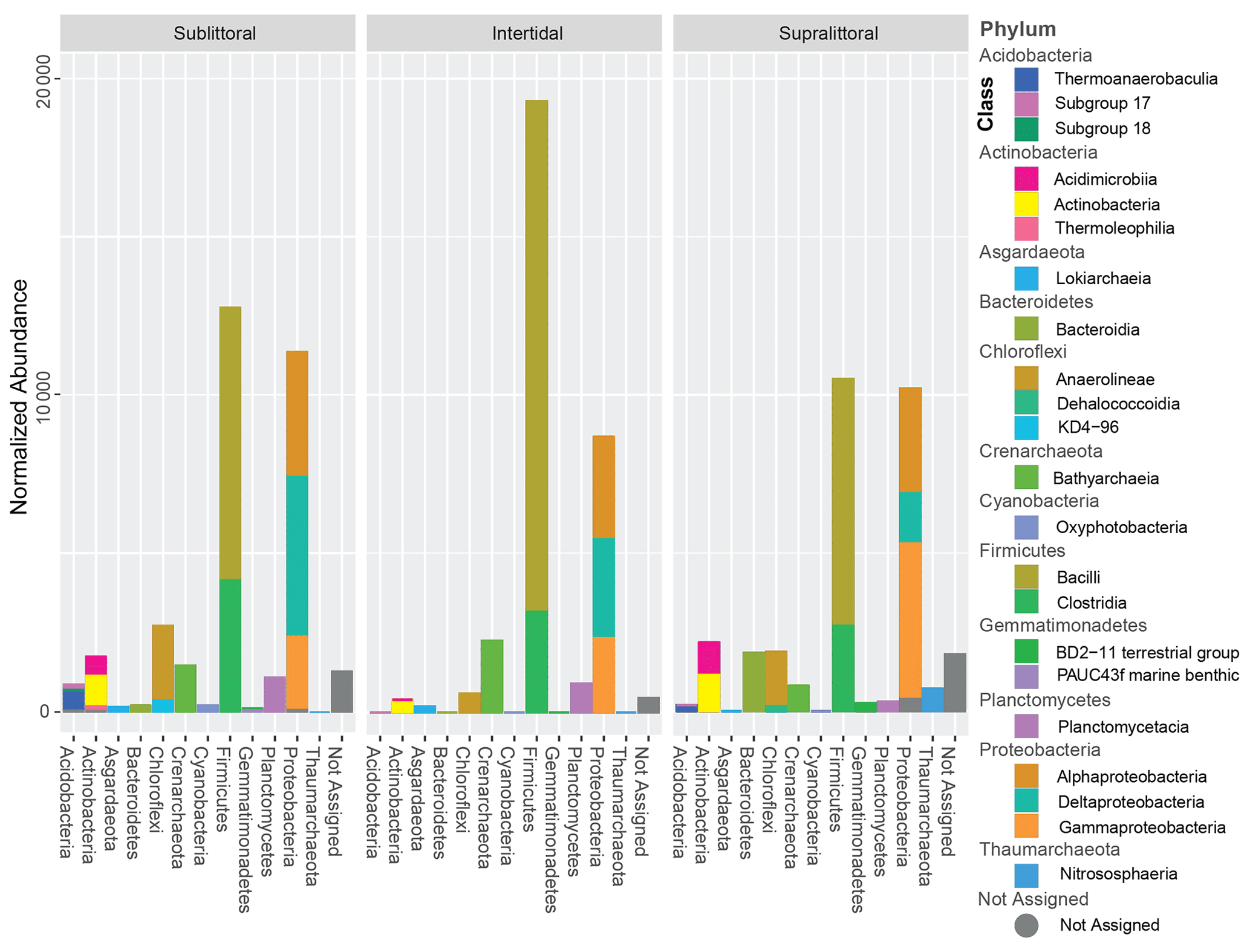
Figure 2Normalized taxonomic abundances from each tidal zone. Taxa identified with an abundance higher than 1 % are shown as stacked bar plots; the horizontal axis is the phylum level while the stacked bars are the class level. Taxa abundance has been normalized by downsampling sequence depth.
2.4.2 Taxonomic assignment, community visualization, and environmental tests
Taxonomic assignment was performed using QIIME 2's naive Bayes scikit-learn classifier (Bokulich et al., 2018) trained using the 16S rRNA gene sequences in the SILVA database (Silva SSU 132) (McDonald et al., 2012) (see Supplement methods, file 1). Additional QIIME 2 visualizations for (1) ASV abundance (Fig. S7 in the Supplement), (2) proportional representation between zones (Supplement file 3), and (3) taxonomy (Supplement file 4) are available as a Supplement files. Phylogenetic reconstruction was carried out in QIIME 2 using the feature classifier trained on 16S rRNA gene sequences. All groups were required to be present within at least two samples with a minimum of three reads each. In one event a taxonomic family was manually relabeled from “Uncultured eukaryote” to “Uncultured Bathyarchaeia” (Figs. 2 and S8 in the Supplement).
The vegan R package (Dixon, 2003; Oksanen et al., 2015; R Core Team, 2019) was used to test correlations between community structure and environmental variables. Distances were calculated using metaMDS (distance used was Bray–Curtis), and environmental variables were fit using envfit (Table S2 in the Supplement). Additionally, we also tried a constrained ordination analysis using vegan's capscale method. However, the linear model fit of the complete data set using this approach did not identify a statistically significant correlation (Fig. S9 in the Supplement).
2.4.3 Functional analysis using PICRUSt2
Functional analysis was performed using PICRUSt2 (Douglas et al., 2019; Barbera et al., 2019; Czech et al., 2020; Louca and Doebeli, 2018; Ye and Doak, 2009) with default settings. As we used 16S rRNA gene amplicon sequencing a crucial limitation must be considered in evaluating our results (Sun et al., 2020), which is that all KEGG ortholog (KO) abundances (and subsequent pathway analysis) are derived from the inferred abundance of genes from the closest reference taxa that matches our supplied taxa generated by QIIME 2. As such, there is the potential for disagreement between the actual organism and the reference we rely on for inference. To address this we use PICRUSt2's nearest sequenced taxon index (NSTI) as a heuristic cutoff in the evaluation of taxon level functional analysis (Sect. 3.3, Table S1 in the Supplement). Although we are using taxonomic families in this study, we will continue to use the NSTI cutoff of 0.15, despite it being derived for species level comparisons. Where relevant families with median NSTI scores above 1 SD are labeled with an asterisk (*), families with an NSTI outside of this range were not included in this analysis. A zone-specific analysis of significant differential abundances was performed using the general linear model method of the ALDEx2 package (see Supplement methods, files 1 and 6). A heatmap of KOs with significant differential abundance between zones was then generated (Fig. 6).
2.4.4 Zone-specific taxonomic and functional enrichment
In order to identify which species were significantly different in abundance in each zone, we performed taxa enrichment analysis (Fig. 4), using a custom Python (Van Rossum and Drake, 2009) script (see Supplement methods, files 1 and 6). To determine which taxa were driving the differences in functional abundance, we calculated taxa-specific KO enrichment (Fig. 4) using the relative functional abundances from PICRUSt2 (see Supplement methods, files 1 and 6). KOs were limited to the KEGG metabolic pathways (ko00030, ko00195, ko00680, ko00710, ko00720, ko00910, ko00920). KOs were deemed enriched in a taxa if that taxa accounted for at least 10 % of the total relative functional abundance for that KO. A taxa was deemed to be enriched for a metabolic pathway (Fig. 4) if it was enriched for three KOs within that pathway. A more detailed analysis of metabolic pathway modules was performed using KEGG Mapper reconstruction to identify instances of taxa enrichment that included complete pathway modules (Fig. 5, Supplement file 7).
2.5 Accessibility
The entire computational workflow is available on GitHub: https://github.com/pspealman/COSantana_2021, last access: 25 March 2021.
The data have been deposited as PRJNA608697 in the NCBI BioProject database: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA608697, last access: 25 March 2021.
3.1 Structural composition of prokaryotic communities
Sequence clustering yielded a total of 2030 ASVs, of which 94.3 % were assigned to Bacteria, 5.3 % were assigned to Archaea, and 0.4 % were not assigned to any prokaryotic kingdom (Fig. S7). All sites combined resulted in 36 phyla, 158 orders, 79 classes, 164 families, 135 genera, and 95 species. Approximately 89 % of all the sequences belonged to 6 phyla: Firmicutes (33.7 %), Proteobacteria (32 %), Chloroflexi (7 %), Planctomycetes (5.9 %), Actinobacteria (5.7 %), and Crenarchaeota (4.4 %). Crenarchaeota, Thaumarchaeota, and Asgardaeota were the representatives of Archaea groups with more than 1 % abundance in the samples (Fig. 2). Taxonomic profiles are broadly similar across the tidal zones at the family level (Fig. S10 in the Supplement). However, several families break from this trend (Supplement file 7), such as the two families of the order Alteromonadales (Fig. S10 – 2. Marinobacteraceae and Pseudoalteromonadaceae) are only abundant in the supralittoral zone. From the class Deltaproteobacteria (Fig. S10 – 9. Desulfarculales, 10. Desulfobacterales, 14. Myxococcales, 23. Syntrophobacterales), the majority of orders and families are either more abundant or only present at the sublittoral zone as well.
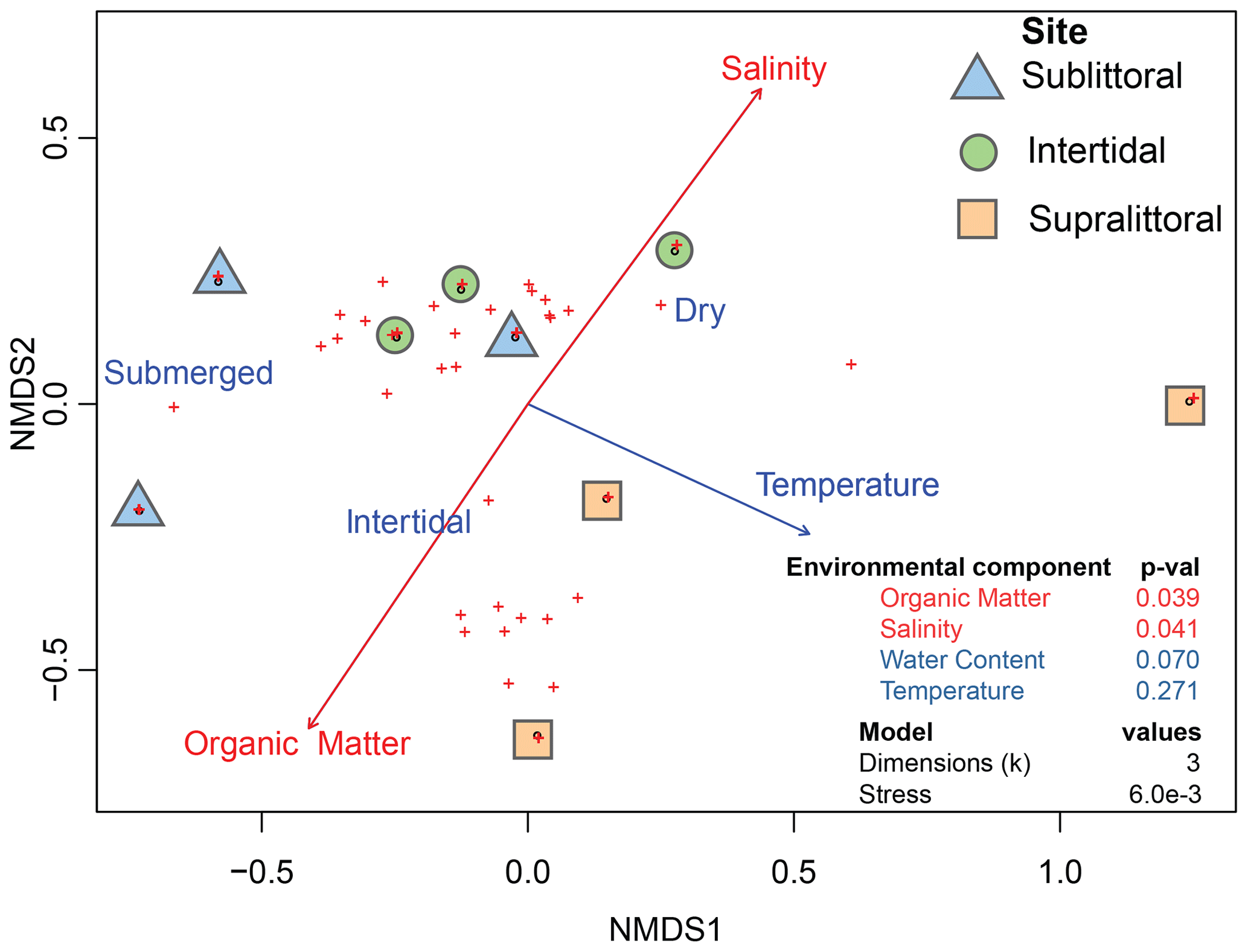
Figure 3Correlation between environmental variables and prokaryotic communities. Dots represent taxonomic abundances per site as plotted by vegan metaMDS using nonmetric multidimensional scaling (NMDS). Arrow length is a representative of the predictor strength of environmental variable vectors, with red arrows having statistical significance as calculated by envfit (p value < = 0.05).
Alpha-diversity analysis showed a trend of the intertidal zone having the lowest prokaryotic diversity (Shannon's index – 3.8), while the sublittoral zone had the highest (Shannon's index – 4.8) and the supralittoral region had intermediary diversity (Shannon's index – 4.2) (Fig. S2). However, these differences between diversity indices were not statistically significant between the tidal zones, suggesting that the communities are similar in terms of biodiversity. Similarly, the beta-diversity analysis revealed no statistical differences in the taxonomic structure of prokaryotic populations between zones (Fig. S5).
3.2 The influence of environmental variables
We also aimed to determine if population structures across mangrove tidal zones correlated with the abiotic environmental variables of salinity, water content, organic matter, and temperature from each tidal zone (Table S2). The results revealed a significant correlation between the prokaryotic populations with salinity and organic matter (Fig. 3, Table S3 in the Supplement). Specifically, increased organic matter was positively correlated with sublittoral population abundances, while increased salinity was positively correlated with supralittoral population abundances. Neither water content nor temperature measures reflect a significant difference in community structure between zones.
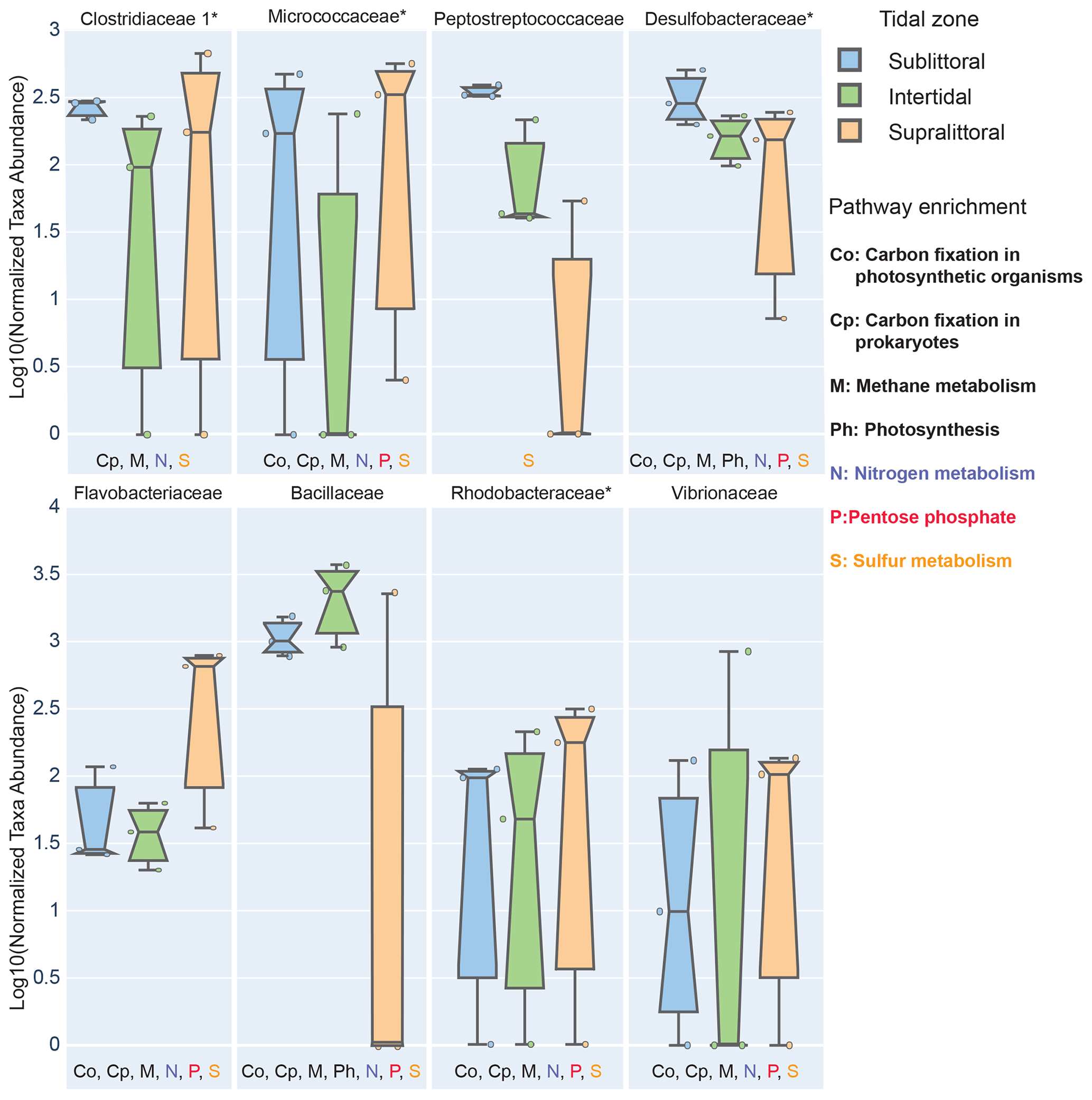
Figure 4Zone-specific measures of taxa showing significant enrichment in sediments of different tidal zones. Here we show prokaryotic families that have significantly different abundances between zones and whose mean effect size exceeded 10 % in at least one zone. To be labeled with a metabolic pathway, taxa were required to have at least 10 % of three KOs in that pathway in any zone. Families with a median NSTI within 1 SD of 0.15 are labeled with *.
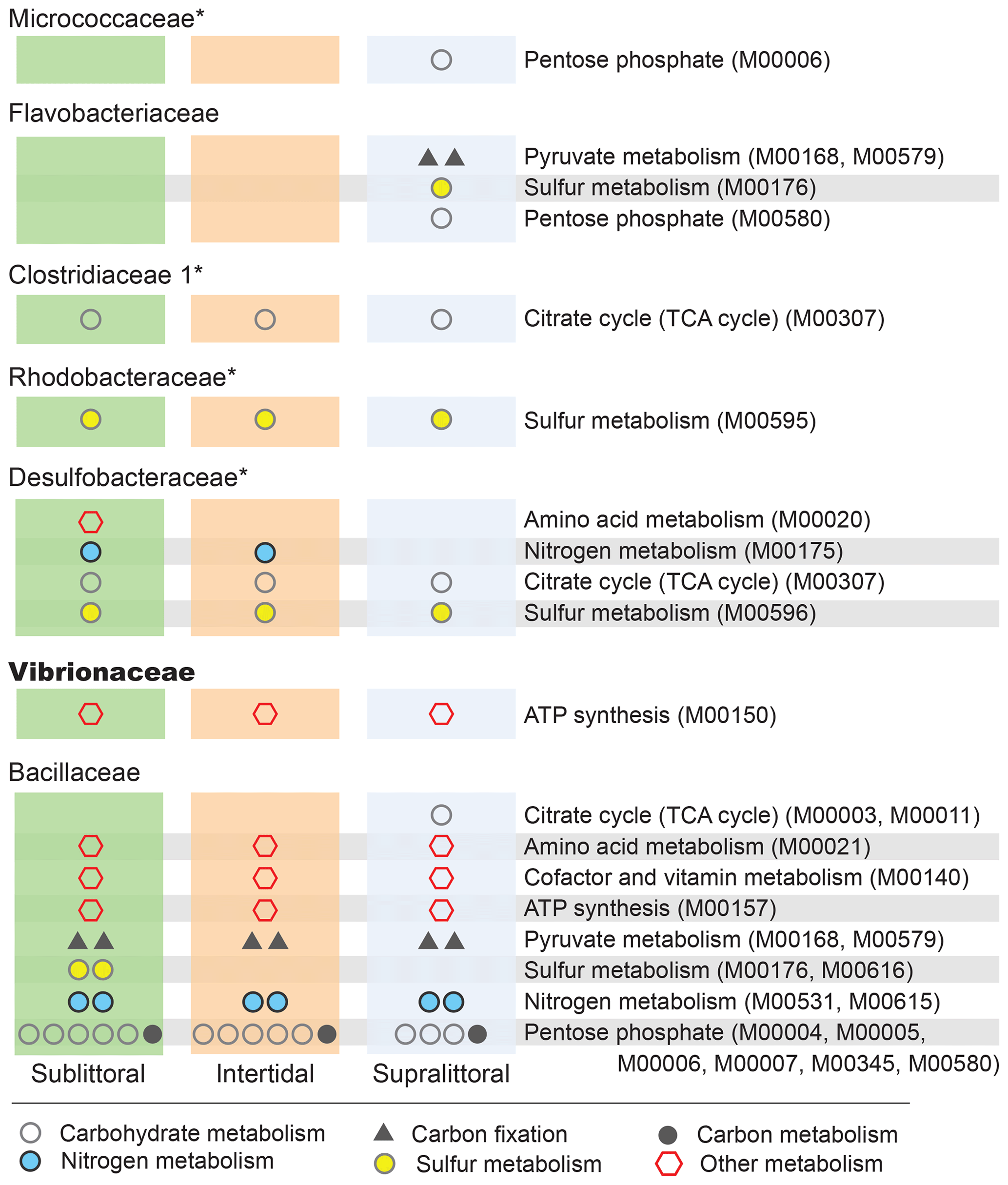
Figure 5Zone-specific enrichment of pathway modules. Here, we show the impact that the differential abundances of major families (Fig. 4) have on potential functional profiles in each zone at the level of pathway modules. To be included here each module must be completely enriched within a single taxon in a single zone, such that the taxa account for at least 10 % of the total potential functional abundance for each KO involved in the pathway. Families with a median NSTI within 1 SD of 0.15 are labeled with *.
3.3 Tidal-zone-specific taxa enrichment and their contributions to metabolic functional potentials
To determine which specific taxa were significantly different in abundance between zones, we performed a zone-specific enrichment test for each taxa (Fig. 4). At the taxonomic level of family, taxa that were a substantial component of the population of at least one zone (at least 10 % of the population) and whose abundance in another zone was significantly different were defined as zone-specific enriched taxa. Additionally, these taxa were also evaluated for their role in metabolic processes; those with enrichment of metabolism-associated KOs (accounting for more than 10 % of the total of a given KO for at least three KOs of a single metabolic pathway) were then labeled for that pathway. We also performed pathway analysis wherein a pathway module would be defined as enriched if the complete set of KOs for that pathway was enriched (Fig. 5).
We found eight families with statistically different population abundances between tidal zones (Bacillaceae, Flavobacteriaceae, Micrococcaceae, Clostridiaceae, Peptostreptococcaceae, Desulfobacteraceae, Rhodobacteraceae, and Vibrionaceae); four of these families (Flavobacteriaceae, Micrococcaceae, Clostridiaceae, and Vibrionaceae) had their lowest population abundances in the intertidal zone, and only one family (Bacillaceae) had its highest abundance in this zone. No archaea satisfied the statistical criteria necessary for inclusion. This dominance by Bacillaceae and reduced abundance of other families is consistent with our previous observations of the intertidal zone having a low diversity. Furthermore, these differences in population abundance can also affect the functional profile of these zones. Each family also contributed substantially (≥ 10 %) to the total potential functional abundance of at least one of the metabolic pathways in one zone (Fig. 4). Of those eight families with the exception of Peptostreptococcaceae, we found each contributed significantly to carbon metabolism, which includes photosynthesis (Ph, two families), carbon fixation in photosynthetic (Co, six families) or heterotrophic (Cp, seven families) organisms, and methane metabolism (M, seven families). All families contributed to sulfur metabolism-associated KOs, and these were mostly associated with assimilatory sulfate reduction (M00176) and dissimilatory sulfate metabolism (M00596). Seven families contributed substantially to nitrogen metabolism-associated KOs. Only six families contributed importantly to the general phosphorus metabolism-associated KOs. Taken together, this suggests that zone-specific taxa enrichment may also contribute to differential metabolic activities in these zones.
We can also analyze KO enrichment at a higher resolution by considering taxa enrichment of metabolic modules (Fig. 5). We find greater taxa-enriched KOs in the sublittoral zone, where 24 out of 29 (82.8 %) taxa-enriched pathways are found, vs. 21 (72.4 %) and 22 (75.9 %), in the intertidal and supralittoral zones, respectively. Notably, Bacillaceae is the most enriched family in each zone and, as such, has the potential to drive a large proportion of functional activity, being enriched in 17 metabolic modules in eight pathways. These include modules from the carbohydrate metabolism pathways (citrate cycle and pentose phosphate), as well as from sulfur and nitrogen metabolism pathways. Notably, some of these are enriched in only one zone, such as sulfur metabolism and the citrate cycle, suggesting that these roles are occupied by other families in these tidal zones.

Figure 6Zone-specific measures of metabolism-associated KEGG orthologs. The heatmaps show the metabolism-associated KOs that have significant differences in functional abundance between zones for carbon, phosphorus, sulfur, and nitrogen pathways. Notably, the majority of KOs (45/54, 83 %) have their highest relative functional abundance in the sublittoral zone, with the intertidal (5/54, 9.3 %) and supralittoral (4/54 7.4 %) zones as near-equal minorities.
3.4 Zone-specific differences in metabolism-associated KEGG orthologs
To determine if there were significant differences in potential metabolic activity between tidal zones, we calculated the potential functional abundance of metabolic KOs for each zone. KOs with significantly different functional abundances between zones are shown in Fig. 6. Overall, the functional profiles showed that the majority of metabolic pathways for the transformations of carbon, nitrogen, phosphorus, and sulfur are more abundant in the sediments of the sublittoral zone. Taken together, the results suggest a positive correlation between biodiversity and potential metabolic functional abundances.
In this work, we extended the study of preserved mangrove areas to characterize the variance in prokaryotic populations within microhabitats defined by the influence of the tidal regime. Previous research has shown that mangrove forests exhibit zonation driven by different biotic and abiotic factors which influence microbial community structure. However, most of this research has been conducted in anthropogenically impacted areas (Pupin and Nahas, 2014; Marcial Gomes et al., 2008; Alzubaidy et al., 2016; Rocha et al., 2016; Ceccon et al., 2019; El-Tarabily, 2002; Imchen et al., 2017; Lin et al., 2019; Zhang et al., 2018; Zhou et al., 2017), thus confounding the makeup of the microbial population structures. Conversely, studies that sought to identify differences induced by pollution and urbanization on mangroves found broad differences in prokaryotic populations in impacted areas compared to preserved mangrove areas (Pupin and Nahas, 2014; Nogueira et al., 2015) but did not study the population differences of distinct microhabitats within mangroves.
Our work shows that the different tidal zones of the Serinhaém estuary's mangrove have similar community structures with distinct functional profiles. We observed a prevalence of Firmicutes in the sediments of all tidal zones, accounting for 34 % of all reads. This differs from previous studies of mangroves, both around the world and in Brazil (Alzubaidy et al., 2016; Andreote et al., 2012; Mendes and Tsai, 2014; Nogueira et al., 2015; Ceccon et al., 2019; Imchen et al., 2017; Cheung et al., 2018; Zhou et al., 2017), which rarely found the relative frequency of Firmicutes to be above 10 % (Figs. S11–S13 in the Supplement). Instead, these studies found Proteobacteria to be the dominant phyla and, while Proteobacteria was also abundant in our samples (32 % of all reads), it never exceeded the abundance of Firmicutes. Notably, Firmicutes were mainly represented by the family Bacillaceae which in our potential functional analysis was an important driver of metabolic cycles. We also compared our observations to coastal marine sediments (Supplemental file 2), and here too, we found Proteobacteria to be the dominant taxa at anthropogenically impacted sites. However, in one study performed at a marine sanctuary there was also the observation of high-abundance Firmicutes (18 %–31 %).
Previous work on contaminated mangroves in Brazil has suggested that Gammaproteobacteria is a bioindicator of an anthropogenically impacted mangrove (Andreote et al., 2012a). Comparing normalized reads across our samples for all Firmicutes and Gammaproteobacteria (Table S4 in the Supplement), we found that the two taxa are significantly negatively correlated (Pearson's R = −0.84; p value = 0.004), consistent with Firmicutes being a bioindicator of a pristine environment. However, Firmicutes were also found at elevated levels at several anthropogenically impacted sites (Torres et al., 2019; Haldar and Nazareth, 2018; Tiralerdpanich et al., 2018). While it could be that their presence is due to the pristine nature of the area, it may also be due to the local climate (Nogueira et al., 2015), an unmeasured environmental factor (Tong et al., 2019), or the presence of another organism such as a non-prokaryote or prokaryote which is difficult to detect using 16S rRNA gene amplicon sequencing (Eloe-Fadrosh et al., 2016; Zhang et al., 2021).
The results show that both salinity and organic matter were significantly correlated with communities in different tidal zones, confirming the hypothesis that changes in physicochemical parameters lead to changes in the prokaryotic communities inhabiting these sediments. It has been previously observed that tidal variations can cause nutrient washout or erosion (Behera et al., 2019; Zhang et al., 2020), which in turn can change the bioavailability patterns for organic matter and essential nutrients along the distinct tidal zones of a mangrove habitat. Salinity levels are also influenced by the tidal variations, and it has been previously determined that the increased salinity as well as the prevalence of humic-like substances can interfere with the accessibility of the microbial community to the sediment organic matter (Wang et al., 2010; Ceccon et al., 2019).
It is important to note that other environmental variables such as granulometry, vegetation, and pollutant distributions may also impact mangrove sediment communities, as observed previously (Peixoto et al., 2011; Colares and Melo, 2013; Rocha et al., 2016). Furthermore, it is also possible that they are influenced by additional biological factors, such as fungi and other eukaryotic microbes (Simões et al., 2015), and plant rhizome contamination (Bennett and Klironomos, 2019; Miller et al., 2019), as observed in previous work (Rocha et al., 2016; Zhang et al., 2017). Thus, our understanding of prokaryotic community structures will be greatly increased if complemented with rhizome and eukaryotic population information.
It is also important to notice that the use of 16S rRNA gene amplicons potentially impairs the identification of some important prokaryotic groups, especially from the domain Archaea, which in some environments can reach 10 % of the total microbial community (Eloe-Fadrosh et al., 2016). Recent research has emphasized how the use of 16S rRNA gene amplicon sequencing can lead to failure in detecting some archaeal populations which can play important roles in nutrient cycling (Zhang et al., 2021). Taken together, however, this work suggests that a variety of environmental factors, potentially driven by tidal cycles, can generate niche variations that influence the structure and function of communities.
Although the differences in diversity indices were not statistically significant, the data show a clear trend of differentiation between zones that suggests that further investigation, with a larger number of samples, would be sufficient to resolve differences with statistical significance. The highest biodiversity was found in the sublittoral mangrove sediments, while the intertidal zone had the lowest biodiversity. The finding of taxonomic groups that were more abundant and/or exclusive to specific zones is also suggestive of the importance of the physical–chemical parameters in shaping the microbial communities. The prevalence of the groups of Deltaproteobacteria in the sublittoral zone, for example, could be associated with the prevalent anaerobic conditions of such sediments, which could result in the selection of specific prokaryotic groups such as sulfate-reducing bacteria (Andreote et al., 2012), as seen with the increased abundance of Desulfobacteraceae, Desulfobulbaceae, and Syntrophaceae.
Our data suggest that the intertidal sediments of mangrove forests have lower prokaryotic diversity than those in the constant environments, such as the supralittoral and sublittoral regions. Models of biodiversity in dynamic environmental regions, such as intertidal zones, have suggested that oscillating environments may support higher biodiversity than that observed in static environments (Brose and Hillebrand, 2016). Furthermore, studies in anthropogenically impacted mangroves have often found substantial diversity in the intertidal zones as well (Lin et al., 2019; Zhang et al., 2018). An alternative model is that the constant shifts in environmental parameters can create harsh environmental conditions that select for resistant taxa thus reducing the relative biodiversity (Statzner and Moss, 2004), which is consistent with our results. Considering other surrounding environments such as the shallow marine sediments (Supplemental file 2), we may expect the diversity to decrease in comparison to the mangrove sediments, potentially with an increase in Proteobacteria, a trend that has been reported in previous studies which found similar groups prevailing in mangrove sediments (Wang et al., 2012; Behera et al., 2019).
Community functional profiling was undertaken using 16S rRNA gene sequences and identified higher metabolic potentials for the cycles of C, N, P, and S in the sublittoral region. The higher abundances of KOs found in the sublittoral zone is possibly due to the greater taxonomic diversity that was also observed for this region. Notably, this result is in agreement with the understanding that many microbiome functions in ecosystems are redundant (Tolkkinen et al., 2020; Barnes et al., 2020). It is important to note that these functional profiles are not the result of metagenomic sequencing; as such it is not necessarily true that all members of the taxa have the same genome as the reference holotype, nor is it necessarily true that these genes would be expressed equally, even if present, and this should best be understood as a measure of potential metabolic activity. Studies that have performed functional profiling using metagenome sequencing on mangrove sediments resulted in comparatively greater resolution (Zhang et al., 2021), although the use of 16S rRNA gene sequencing has been proved to provide comparable results for the recovery of general patterns of pathway abundances (Jovel et al., 2016).
Generally, diverse communities with organisms possessing redundant metabolic functions may be more stable against environmental perturbations, as the organisms will respond differently to stressors, increasing the likelihood of survival of some taxa (Girvan et al., 2005). Thus, the identification of the intertidal zone as having the lowest diversity suggests it may be in a more precarious position. This concern is amplified as the water itself is frequently the carrier of contamination, both from rivers, as is the case for urban waste (Yunus et al., 2011), and from the oceans through the tides, as the case with oil spill contamination (Cabral et al., 2016). Thus, it is important to consider that different parts of the mangrove tidal zone could be exposed to different levels of contamination and that this could, in turn, affect the organisms in a zone-specific manner.
The identification of enriched taxa with similar functional potentials for the diverse nutrient metabolisms is in agreement with previous observations of the microbial diversity in mangrove sediments (Rocha et al., 2016; Ceccon et al., 2019; Cabral et al., 2016; Mendes and Tsai, 2014; Zhao and Bajic, 2015). Conceptually, one could interpret that this higher-order redundant biodiversity could grant the microbiome a higher level of resilience when challenged by environmental perturbation, as organisms with similar metabolic pathways have the potential to maintain the ecological functions even if some taxa are lost.
In conclusion, the results suggest that communities with distinct diversity and functional profiles occupy the sediments of the different mangrove tidal zones. This variance is significantly associated with the abiotic environmental variables of salinity and organic matter. Our functional potential analysis suggests there could be significant variation in metabolic potentials between tidal zones. We observed that the intertidal region, the most biophysically dynamic zone in the mangrove forest, presents the least biodiversity. One possible explanation for this is that the dynamic, cyclic, tidal environment is itself harsher than that found in the other tidal zones. This study of pristine mangrove tidal zones suggests that some microhabitats may be more sensitive to anthropogenic impact than others. Preservation, conservation, and remediation measures of mangroves should take into account the fragility of the intertidal zone and the increased threat this zone faces from soluble pollutants, such as urban sewage, fertilizer, and pesticide runoff, as well as from buoyant pollution such as plastics and oil spills. However, it remains an open question if other microhabitats, such as the sublittoral zone, have greater resilience to environmental perturbations given their greater abundance of taxonomic diversity and metabolic function. Conceptually, this property of a dynamic environment defining a selective niche, similarly to a physical barrier, is worthy of further study. Further exploration of whether different groups of organisms found to have metabolic potentials are actively participating in the element cycles in these mangrove sediments will also benefit the field.
All custom code used in the analysis is available for download through GitHub (https://github.com/pspealman/COSantana_2021, last access: 25 March 2021) and as an archived repository (https://doi.org/10.5281/zenodo.4637755; Spealman et al., 2021a; last access: 26 March 2021).
All sequencing data have been deposited as PRJNA608697 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA608697, Santana, 2020, last access: 25 March 2021). Supplement, intermediate, and metadata files are also available for download through an archived repository (https://doi.org/10.5061/dryad.gf1vhhmkz; Spealman et al., 2021b; last access: 26 March 2021).
The supplement related to this article is available online at: https://doi.org/10.5194/bg-18-2259-2021-supplement.
COdS worked on the conceptualization, formal analysis, investigation, visualization, writing, and the original draft preparation of the manuscript. PS made contributions to the formal statistical and genomic analysis, visualization, and writing and to the corrections of the manuscript. VMMM contributed with necessary resources for genomic and data analysis. DG provided resources for statistical and genomic analysis while supervising results and contributing with conceptualization and writing of the original draft of the manuscript. TBdJ worked on the conceptualization of the research and funding acquisition and provided resources for fieldwork and laboratory analysis. FAC contributed to the steps of conceptualization of the study and project administration and provided resources for laboratory analysis.
The authors declare that they have no conflict of interest.
The authors would like to thank the sequencing facility of the Microbial Ecology and Biotechnology Laboratory (Lembiotech).
This work was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil, Finance Code 001 (https://www.capes.gov.br/, last access: 25 March 2021), and by the Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB) (http://www.fapesb.ba.gov.br/, last access: 25 March 2021) (grant no. PNE0021/2014). Additional funding was provided by the National Science Foundation (NSF) (grant no. MCB1818234).
This paper was edited by Denise Akob and reviewed by three anonymous referees.
Allard, S. M., Costa, M. T., Bulseco, A. N., Helfer, V., Wilkins, L. G. E., Hassenrück, C., Zengler, K., Zimmer, M., Erazo, N., Mazza Rodrigues, J. L., Duke, N., Melo, V. M. M., Vanwonterghem, I., Junca, H., Makonde, H. M., Jiménez, D. J., Tavares, T. C. L., Fusi, M., Daffonchio, D., Duarte, C. M., Peixoto, R. S., Rosado, A. S., Gilbert, J. A., and Bowman, J.: Introducing the Mangrove Microbiome Initiative: Identifying Microbial Research Priorities and Approaches To Better Understand, Protect, and Rehabilitate Mangrove Ecosystems, mSystems, 5, e00658-20, https://doi.org/10.1128/msystems.00658-20, 2020.
Alongi, D. M.: Bacterial productivity and microbial biomass in tropical mangrove sediments, Microb. Ecol., 15, 59–79, https://doi.org/10.1007/BF02012952, 1988.
Alzubaidy, H., Essack, M., Malas, T. B., Bokhari, A., Motwalli, O., Kamanu, F. K., Jamhor, S. A., Mokhtar, N. A., Antunes, A., Simões, M. F., Alam, I., Bougouffa, S., Lafi, F. F., Bajic, V. B., and Archer, J. A. C.: Rhizosphere microbiome metagenomics of gray mangroves (Avicennia marina) in the Red Sea, Gene, 576, 626–636, https://doi.org/10.1016/j.gene.2015.10.032, 2016.
Andreote, F. D., Jiménez, D. J., Chaves, D., Dias, A. C. F., Luvizotto, D. M., Dini-Andreote, F., Fasanella, C. C., Lopez, M. V., Baena, S., Taketani, R. G., and de Melo, I. S.: The microbiome of Brazilian mangrove sediments as revealed by metagenomics, PLoS One, 7, e38600, https://doi.org/10.1371/journal.pone.0038600, 2012.
Azzoni, R., Nizzoli, D., Bartoli, M., Christian, R. R., and Viaroli, P.: Factors Controlling Benthic Biogeochemistry in Urbanized Coastal Systems: an Example from Venice (Italy), Estuaries Coasts, 38, 1016–1031, https://doi.org/10.1007/s12237-014-9882-6, 2015.
Barbera, P., Kozlov, A. M., Czech, L., Morel, B., Darriba, D., Flouri, T., and Stamatakis, A.: EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences, Syst. Biol., 68, 365–369, https://doi.org/10.1093/sysbio/syy054, 2019.
Barnes, E. M., Carter, E. L., and Lewis, J. D.: Predicting Microbiome Function Across Space Is Confounded by Strain-Level Differences and Functional Redundancy Across Taxa, Front. Microbiol., 11, 101, https://doi.org/10.3389/fmicb.2020.00101, 2020.
Behera, P., Mohapatra, M., Kim, J. Y., Adhya, T. K., Pattnaik, A. K., and Rastogi, G.: Spatial and temporal heterogeneity in the structure and function of sediment bacterial communities of a tropical mangrove forest, Environ. Sci. Pollut. R., 26, 3893–3908, https://doi.org/10.1007/s11356-018-3927-5, 2019.
Bennett, J. A. and Klironomos, J.: Mechanisms of plant-soil feedback: interactions among biotic and abiotic drivers, New Phytol., 222, 91–96, https://doi.org/10.1111/nph.15603, 2019.
Boehm, A. B., Yamahara, K. M., and Sassoubre, L. M.: Diversity and transport of microorganisms in intertidal sands of the California coast, Appl. Environ. Microb., 80, 3943–3951, https://doi.org/10.1128/AEM.00513-14, 2014.
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., Huttley, G. A., and Gregory Caporaso, J.: Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2's q2-feature-classifier plugin, Microbiome, 6, 90, https://doi.org/10.1186/s40168-018-0470-z, 2018.
Bolger, A. M., Lohse, M., and Usadel, B.: Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170, 2014.
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., Alexander, H., Alm, E. J., Arumugam, M., Asnicar, F., Bai, Y., Bisanz, J. E., Bittinger, K., Brejnrod, A., Brislawn, C. J., Brown, C. T., Callahan, B. J., Caraballo-Rodríguez, A. M., Chase, J., Cope, E. K., Da Silva, R., Diener, 70 C., Dorrestein, P. C., Douglas, G. M., Durall, D. M., Duvallet, C., Edwardson, C. F., Ernst, M., Estaki, M., Fouquier, J., Gauglitz, J. M., Gibbons, S. M., Gibson, D. L., Gonzalez, A., Gorlick, K., Guo, J., Hillmann, B., Holmes, S., Holste, H., Huttenhower, C., Huttley, G. A., Janssen, S., Jarmusch, A. K., Jiang, L., Kaehler, B. D., Kang, K. B., Keefe, C. R., Keim, P., Kelley, S. T., Knights, D., Koester, I., Kosciolek, T., Kreps, J., Langille, M. G. I., Lee, J., Ley, R., Liu, Y.-X., Loftfield, E., Lozupone, C., Maher, M., Marotz, C., Martin, B. D., McDonald, D., McIver, L. J., Melnik, A. V., Metcalf, J. L., Morgan, S. C., Morton, J. T., Naimey, A. T., Navas-Molina, J. A., Nothias, L. F., Orchanian, S. B., Pearson, T., Peoples, S. L., Petras, D., Preuss, M. L., Pruesse, E., Rasmussen, L. B., Rivers, A., Robeson, M. S., 2nd, Rosenthal, P., Segata, N., Shaffer, M., Shiffer, A., Sinha, R., Song, S. J., Spear, J. R., Swafford, A. D., Thompson, L. R., Torres, P. J., Trinh, P., Tripathi, A., Turnbaugh, P. J., Ul-Hasan, S., van der Hooft, J. J. J., Vargas, F., Vázquez-Baeza, Y., Vogtmann, E., von Hippel, M., Walters, W., Wan, Y., Wang, M., Warren, J., Weber, K. C., Williamson, C. H. D., Willis, A. D., Xu, Z. Z., Zaneveld, J. R, Zhang, Y., Zhu, Q., Knight, R., and Caporaso, J. G.: Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2, Nat. Biotechnol., 37, 852–857, https://doi.org/10.1038/s41587-019-0209-9, 2019.
Brooks, T., Tobias, J., and Balmford, A.: Deforestation and bird extinctions in the Atlantic forest, Anim. Conserv., 2, 211–222, https://doi.org/10.1111/j.1469-1795.1999.tb00067.x, 1999.
Brose, U. and Hillebrand, H.: Biodiversity and ecosystem functioning in dynamic landscapes, Philos. T. Roy. Soc. B, 371, 20150267, https://doi.org/10.1098/rstb.2015.0267, 2016.
Cabral, L., Júnior, G. V. L., Pereira de Sousa, S. T., Dias, A. C. F., Lira Cadete, L., Andreote, F. D., Hess, M., and de Oliveira, V. M.: Anthropogenic impact on mangrove sediments triggers differential responses in the heavy metals and antibiotic resistomes of microbial communities, Environ. Pollut., 216, 460–469, https://doi.org/10.1016/j.envpol.2016.05.078, 2016.
Cabral, L., Pereira de Sousa, S. T., Júnior, G. V. L., Hawley, E., Andreote, F. D., Hess, M., and de Oliveira, V. M.: Microbial functional responses to long-term anthropogenic impact in mangrove soils, Ecotox. Environ. Safe., 160, 231–239, https://doi.org/10.1016/j.ecoenv.2018.04.050, 2018.
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P.: DADA2: High-resolution sample inference from Illumina amplicon data, Nat. Methods, 13, 581–583, https://doi.org/10.1038/nmeth.3869, 2016.
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., Fierer, N., and Knight, R.: Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample, P. Natl. Acad. Sci. USA, 108 Suppl 1, 4516–4522, https://doi.org/10.1073/pnas.1000080107, 2011.
Ceccon, D. M., Faoro, H., da Cunha Lana, P., de Souza, E. M., and Pedrosa, F. O.: Metataxonomic and metagenomic analysis of mangrove microbiomes reveals community patterns driven by salinity and pH gradients in Paranaguá Bay, Brazil, Sci. Total Environ., 694, 133609, https://doi.org/10.1016/j.scitotenv.2019.133609, 2019.
Cheung, M. K., Wong, C. K., Chu, K. H., and Kwan, H. S.: Community Structure, Dynamics and Interactions of Bacteria, Archaea and Fungi in Subtropical Coastal Wetland Sediments, Sci. Rep., 8, 14397, https://doi.org/10.1038/s41598-018-32529-5, 2018.
Colares, G. B. and Melo, V. M. M.: Relating microbial community structure and environmental variables in mangrove sediments inside Rhizophora mangle L. habitats, Appl. Soil Ecol., 64, 171–177, https://doi.org/10.1016/j.apsoil.2012.12.004, 2013.
Coldren, G. A., Langley, J. A., Feller, I. C., and Chapman, S. K.: Warming accelerates mangrove expansion and surface elevation gain in a subtropical wetland, J. Ecol., 107, 79–90, https://doi.org/10.1111/1365-2745.13049, 2019.
Corrêa-Gomes, L. C., Dominguez, J. M. L., Barbosa, J. S. F., and da Silva, I. C.: PADRÕES DE ORIENTAÇÃO DOS CAMPOS DE TENSÃO, ESTRUTURAS, HERANÇA DO EMBASAMENTO E EVOLUÇÃO TECTÔNICA DAS BACIAS DE CAMAMÚ E PORÇÃO SUL DO RECÔNCAVO, COSTA DO DENDÊ, BAHIA, Revista Brasileira de Geociências, 35, 117–128, https://doi.org/10.25249/0375-7536.200535s4117128, 2005.
Czech, L., Barbera, P., and Stamatakis, A.: Genesis and Gappa: processing, analyzing and visualizing phylogenetic (placement) data, Bioinformatics, 36, 3263–3265, https://doi.org/10.1093/bioinformatics/btaa070, 2020.
da Silva Pereira, M.: Espacialização geoquímica dos sedimentos do estuário do Rio Serinhaém, Baixo Sul da Bahia – Brasil, PROGRAMA DE PÓS-GRADUAÇÃO EM MODELAGEM EM CIÊNCIAS DA TERRA E DO AMBIENTE – PPGM, Universidade Estadual de Feira de Santana, 2016.
Ditt, E., Zysman, N., da Cunha, R. S. and da Rocha, R. B.: Conservação Da Biodiversidade Por Meio Da Atividade Extrativista Em Comunidades Quilombolas, Revista Brasileira de Ciências Ambientais, 27, 1–15, 2013.
Dixon, P.: VEGAN, a package of R functions for community ecology, J. Veg. Sci., 14, 927–930, https://doi.org/10.1658/1100-9233(2003)014[0927:vaporf]2.0.co;2, 2003.
Douglas, G. M., Maffei, V. J., Zaneveld, J., Yurgel, S. N., Brown, J. R., Taylor, C. M., Huttenhower, C., and Langille, M. G. I.: PICRUSt2: An improved and extensible approach for metagenome inference, Bioinformatics, 497, 1–42, 2019.
Eloe-Fadrosh, E. A., Ivanova, N. N., Woyke, T., and Kyrpides, N. C.: Metagenomics uncovers gaps in amplicon-based detection of microbial diversity, Nat. Microbiol., 1, 15032, https://doi.org/10.1038/nmicrobiol.2015.32, 2016.
El-Tarabily, K. A.: Total microbial activity and microbial composition of a mangrove sediment are reduced by oil pollution at a site in the Arabian Gulf, Can. J. Microbiol., 48, 176–182, https://doi.org/10.1139/w01-140, 2002.
Giri, C., Ochieng, E., Tieszen, L. L., Zhu, Z., Singh, A., Loveland, T., Masek, J., and Duke, N.: Status and distribution of mangrove forests of the world using earth observation satellite data: Status and distributions of global mangroves, Global Ecol. Biogeogr., 20, 154–159, https://doi.org/10.1111/j.1466-8238.2010.00584.x, 2011.
Girvan, M. S., Campbell, C. D., Killham, K., Prosser, J. I., and Glover, L. A.: Bacterial diversity promotes community stability and functional resilience after perturbation, Environ. Microbiol., 7, 301–313, https://doi.org/10.1111/j.1462-2920.2005.00695.x, 2005.
Haldar, S. and Nazareth, S. W.: Taxonomic diversity of bacteria from mangrove sediments of Goa: metagenomic and functional analysis, Biotech., 8, 436, https://doi.org/10.1007/s13205-018-1441-6, 2018.
Holguin, G., Vazquez, P., and Bashan, Y.: The role of sediment microorganisms in the productivity, conservation, and rehabilitation of mangrove ecosystems: an overview, Biol. Fert. Soils, 33, 265–278, https://doi.org/10.1007/s003740000319, 2001.
Imchen, M., Kumavath, R., Barh, D., Azevedo, V., Ghosh, P., Viana, M., and Wattam, A. R.: Searching for signatures across microbial communities: Metagenomic analysis of soil samples from mangrove and other ecosystems, Sci. Rep.-UK, 7, 8859, https://doi.org/10.1038/s41598-017-09254-6, 2017.
Jiang, X.-T., Peng, X., Deng, G.-H., Sheng, H.-F., Wang, Y., Zhou, H.-W., and Tam, N. F.-Y.: Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland, Microb. Ecol., 66, 96–104, https://doi.org/10.1007/s00248-013-0238-8, 2013.
Jovel, J., Patterson, J., Wang, W., Hotte, N., O'Keefe, S., Mitchel, T., Perry, T., Kao, D., Mason, A. L., Madsen, K. L., and Wong, G. K.-S.: Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics, Front. Microbiol., 7, 459, https://doi.org/10.3389/fmicb.2016.00459, 2016.
Lin, X., Hetharua, B., Lin, L., Xu, H., Zheng, T., He, Z., and Tian, Y.: Mangrove Sediment Microbiome: Adaptive Microbial Assemblages and Their Routed Biogeochemical Processes in Yunxiao Mangrove National Nature Reserve, China, Microb. Ecol., 78, 57–69, https://doi.org/10.1007/s00248-018-1261-6, 2019.
Lopes, N. S.: Análise da paisagem com base na fragmentação – caso APA do Pratigi, baixo sul da Bahia, Brasil, Revista Eletrônica do Prodema, 6, 53–67, 2011.
Louca, S. and Doebeli, M.: Efficient comparative phylogenetics on large trees, Bioinformatics, 34, 1053–1055, https://doi.org/10.1093/bioinformatics/btx701, 2018.
Luis, P., Saint-Genis, G., Vallon, L., Bourgeois, C., Bruto, M., Marchand, C., Record, E., and Hugoni, M.: Contrasted ecological niches shape fungal and prokaryotic community structure in mangroves sediments, Environ. Microbiol., 21, 1407–1424, https://doi.org/10.1111/1462-2920.14571, 2019.
Lv, X., Ma, B., Yu, J., Chang, S. X., Xu, J., Li, Y., Wang, G., Han, G., Bo, G., and Chu, X.: Bacterial community structure and function shift along a successional series of tidal flats in the Yellow River Delta, Sci. Rep.-UK, 6, 36550, https://doi.org/10.1038/srep36550, 2016.
Marcial Gomes, N. C., Borges, L. R., Paranhos, R., Pinto, F. N., Mendonça-Hagler, L. C. S., and Smalla, K.: Exploring the diversity of bacterial communities in sediments of urban mangrove forests, FEMS Microbiol. Ecol., 66, 96–109, https://doi.org/10.1111/j.1574-6941.2008.00519.x, 2008.
Marcos, M. S., Barboza, A. D., Keijzer, R. M., and Laanbroek, H. J.: Tide as Steering Factor in Structuring Archaeal and Bacterial Ammonia-Oxidizing Communities in Mangrove Forest Soils Dominated by Avicennia germinans and Rhizophora mangle, Microb. Ecol., 75, 997–1008, https://doi.org/10.1007/s00248-017-1091-y, 2018.
Mascarenhas, R. B., Aragão, I. R., Reis, P., and de Jesus Bomfim, T.: ANÁLISE DE METAIS-TRAÇOS EM SEDIMENTOS DA APA DO PRATIGI, BAHIA, Sitientibus, 53, 32–37, https://doi.org/10.13102/sitientibus.v0i53.4467, 2019.
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., Andersen, G. L., Knight, R., and Hugenholtz, P.: An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea, ISME J., 6, 610–618, https://doi.org/10.1038/ismej.2011.139, 2012.
Mendes, L. W. and Tsai, S. M.: Variations of Bacterial Community Structure and Composition in Mangrove Sediment at Different Depths in Southeastern Brazil, Diversity, 6, 827–843, https://doi.org/10.3390/d6040827, 2014.
Miller, E. C., Perron, G. G., and Collins, C. D.: Plant-driven changes in soil microbial communities influence seed germination through negative feedbacks, Ecol. Evol., 9, 9298–9311, https://doi.org/10.1002/ece3.5476, 2019.
MMA, Ministério do Meio Ambiente: Plano de Manejo da APA do Pratigi – Encarte II Zoneamento e Plano de Gestao, available at: http://www.inema.ba.gov.br/wp-content/uploads/2011/09/PM_APA_Pratigi_Encarte-II.pdf (last access: 25 March 2021), 2004.
Musat, N., Werner, U., Knittel, K., Kolb, S., Dodenhof, T., van Beusekom, J. E. E., de Beer, D., Dubilier, N., and Amann, R.: Microbial community structure of sandy intertidal sediments in the North Sea, Sylt–Rømø Basin, Wadden Sea, Syst. Appl. Microbiol., 29, 333–348, https://doi.org/10.1016/j.syapm.2005.12.006, 2006.
Nelson, D. W. and Sommers, L. E.: Total Carbon, Organic Carbon, and Organic Matter: Part 3 Chemical Methods, in: Methods of Soil Analysis, edited by: Sparks, D. L., Page, A. L., Helmke, P. A., Loeppert, R. H., Soltanpour, P. N., Tabatabai, M. A., Johnston, C. T., and Sumner, M. E., Soil Science Society of America, American Society of Agronomy, Madison, WI, USA, 961–1010, 1996.
Nogueira, V. L. R., Rocha, L. L., Colares, G. B., Angelim, A. L., Normando, L. R. O., Cantão, M. E., Agnez-Lima, L. F., Andreote, F. D., and Melo, V. M. M.: Microbiomes and potential metabolic pathways of pristine and anthropized Brazilian mangroves, Regional Studies in Marine Science, 2, 56–64, https://doi.org/10.1016/j.rsma.2015.08.008, 2015.
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R. B., Simpson, G. L., Solymos, P., Stevens, M. H. H., and Wagner, H.: Vegan: community ecology package. R package vegan, vers. 2.2-1, R package version 2. 2, 1, available at: https://apps.worldagroforestry.org/publication/vegan-community-ecology-package-r-package-vegan-vers-22-1, (last access: 25 March 2021), 2015.
Parada, A. E., Needham, D. M., and Fuhrman, J. A.: Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples, Environ. Microbiol., 18, 1403–1414, https://doi.org/10.1111/1462-2920.13023, 2016.
Peixoto, R., Chaer, G. M., Carmo, F. L., Araújo, F. V., Paes, J. E., Volpon, A., Santiago, G. A., and Rosado, A. S.: Bacterial communities reflect the spatial variation in pollutant levels in Brazilian mangrove sediment, A. Van Leeuw. J. Microb., 99, 341–354, https://doi.org/10.1007/s10482-010-9499-0, 2011.
Pupin, B. and Nahas, E.: Microbial populations and activities of mangrove, restinga and Atlantic forest soils from Cardoso Island, Brazil, J. Appl. Microbiol., 116, 851–864, https://doi.org/10.1111/jam.12413, 2014.
R Core Team: R: A Language and Environment for Statistical Computing, available at: https://www.R-project.org/, (last access: 25 March 2021), 2019.
Reis, C. R. G., Nardoto, G. B., and Oliveira, R. S.: Global overview on nitrogen dynamics in mangroves and consequences of increasing nitrogen availability for these systems, Plant Soil, 410, 1–19, https://doi.org/10.1007/s11104-016-3123-7, 2017.
Rocha, L. L., Colares, G. B., Nogueira, V. L. R., Paes, F. A., and Melo, V. M. M.: Distinct Habitats Select Particular Bacterial Communities in Mangrove Sediments, Int. J. Microbiol., 2016, 3435809, https://doi.org/10.1155/2016/3435809, 2016.
Saintilan, N., Khan, N. S., Ashe, E., Kelleway, J. J., Rogers, K., Woodroffe, C. D., and Horton, B. P.: Thresholds of mangrove survival under rapid sea level rise, Science, 368, 1118–1121, https://doi.org/10.1126/science.aba2656, 2020.
Santana, C. O.: Metagenomics within mangrove forest tidal zone, NCBI [data set], available at: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA608697/ (last access 25 March 2021), 2020.
Shiau, Y.-J. and Chiu, C.-Y.: Biogeochemical Processes of C and N in the Soil of Mangrove Forest Ecosystems, Forests, 11, 492, https://doi.org/10.3390/f11050492, 2020.
Simões, M. F., Antunes, A., Ottoni, C. A., Amini, M. S., Alam, I., Alzubaidy, H., Mokhtar, N.-A., Archer, J. A. C., and Bajic, V. B.: Soil and rhizosphere associated fungi in gray mangroves (Avicennia marina) from the Red Sea—a metagenomic approach, Genomics Proteomics Bioinformatics, 13, 310–320, available at: https://www.sciencedirect.com/science/article/pii/S1672022915001382 (last access: 25 March 2021), 2015.
Spealman, P., Santana, C., Gresham, D., Melo, V. M. M., Bomfim de Jesus, T., and Chinalia, F. A.: Effects of tidal influence on the structure and function of prokaryotic communities in the sediments of a pristine Brazilian mangrove, Zenodo, https://doi.org/10.5281/zenodo.4637755, 2021a.
Spealman, P., Santana, C., Gresham, D., Melo, V. M. M., Bomfim de Jesus, T., and Chinalia, F. A.: Effects of tidal influence on the structure and function of prokaryotic communities in the sediments of a pristine Brazilian mangrove, DRYAD, Supplement, intermediate, and metadata files, https://doi.org/10.5061/dryad.gf1vhhmkz, 2021b.
Statzner, B. and Moss, B.: Linking ecological function, biodiversity and habitat: a mini-review focusing on older ecological literature, Basic Appl. Ecol., 5, 97–106, https://doi.org/10.1078/1439-1791-00221, 2004.
Sun, S., Jones, R. B., and Fodor, A. A.: Inference-based accuracy of metagenome prediction tools varies across sample types and functional categories, Microbiome, 8, 46, https://doi.org/10.1186/s40168-020-00815-y, 2020.
Tiralerdpanich, P., Sonthiphand, P., Luepromchai, E., Pinyakong, O., and Pokethitiyook, P.: Potential microbial consortium involved in the biodegradation of diesel, hexadecane and phenanthrene in mangrove sediment explored by metagenomics analysis, Mar. Pollut. Bull., 133, 595–605, https://doi.org/10.1016/j.marpolbul.2018.06.015, 2018.
Tolkkinen, M. J., Heino, J., Ahonen, S. H. K., Lehosmaa, K., and Mykrä, H.: Streams and riparian forests depend on each other: A review with a special focus on microbes, Forest Ecol. Manag., 462, 117962, https://doi.org/10.1016/j.foreco.2020.117962, 2020.
Tong, T., Li, R., Wu, S., and Xie, S.: The distribution of sediment bacterial community in mangroves across China was governed by geographic location and eutrophication, Mar. Pollut. Bull., 140, 198–203, https://doi.org/10.1016/j.marpolbul.2019.01.046, 2019.
Torres, G. G., Figueroa-Galvis, I., Muñoz-García, A., Polanía, J., and Vanegas, J.: Potential bacterial bioindicators of urban pollution in mangroves, Environ. Pollut., 255, 113293, https://doi.org/10.1016/j.envpol.2019.113293, 2019.
Van Rossum, G. and Drake, F. L.: Python 3 Reference Manual, CreateSpace Independent Publishing Platform, Scotts Valley, CA, https://doi.org/10.5555/1593511, 2009.
Wang, Y., Sheng, H.-F., He, Y., Wu, J.-Y., Jiang, Y.-X., Tam, N. F.-Y., and Zhou, H.-W.: Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags, Appl. Environ. Microbiol., 78, 8264–8271, https://doi.org/10.1128/AEM.01821-12, 2012.
Wang, Z.-Y., Yuan-Zheng, X. I. N., Dong-Mei, G. A. O., Feng-Min, L. I., Morgan, J., and Xing, B.-S.: Microbial Community Characteristics in a Degraded Wetland of the Yellow River Delta, Pedosphere, 20, 466–478, https://doi.org/10.1016/s1002-0160(10)60036-7, 2010.
Ye, Y. and Doak, T. G.: A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes, PLoS Comput. Biol., 5, e1000465, https://doi.org/10.1371/journal.pcbi.1000465, 2009.
Yun, J., Deng, Y., and Zhang, H.: Anthropogenic protection alters the microbiome in intertidal mangrove wetlands in Hainan Island, Appl. Microbiol. Biot., 101, 6241–6252, https://doi.org/10.1007/s00253-017-8342-1, 2017.
Yunus, K., Yusuf, N. M., Shazili, N. A. M., Chuan, O. M., Saad, S., Chowdhury, A. J. K., and Bidai, J.: Heavy metal concentration in the surface sediment of Tanjung Lumpur mangrove forest, Kuantan, Malaysia, Sains Malays., 40, 89–92, available at: http://journalarticle.ukm.my/2457/1/02_Kamaruzzaman.pdf (last access: 25 March 2021), 2011.
Zhang, H., Liu, F., Zheng, S., Chen, L., Zhang, X., and Gong, J.: The differentiation of iron-reducing bacterial community and iron-reduction activity between riverine and marine sediments in the Yellow River estuary, Marine Life Science & Technology, 2, 87–96, https://doi.org/10.1007/s42995-019-00001-6, 2020.
Zhang, J.-W., Dong, H.-P., Hou, L.-J., Liu, Y., Ou, Y.-F., Zheng, Y.-L., Han, P., Liang, X., Yin, G.-Y., Wu, D.-M., Liu, M., and Li, M.: Newly discovered Asgard archaea Hermodarchaeota potentially degrade alkanes and aromatics via alkyl/benzyl-succinate synthase and benzoyl-CoA pathway, ISME J., https://doi.org/10.1038/s41396-020-00890-x, 2021.
Zhang, X., Hu, B. X., Ren, H., and Zhang, J.: Composition and functional diversity of microbial community across a mangrove-inhabited mudflat as revealed by 16S rDNA gene sequences, Sci. Total Environ., 633, 518–528, https://doi.org/10.1016/j.scitotenv.2018.03.158, 2018.
Zhang, Y., Yang, Q., Ling, J., Van Nostrand, J. D., Shi, Z., Zhou, J., and Dong, J.: Diversity and Structure of Diazotrophic Communities in Mangrove Rhizosphere, Revealed by High-Throughput Sequencing, Front. Microbiol., 8, 2032, https://doi.org/10.3389/fmicb.2017.02032, 2017.
Zhao, F. and Bajic, V. B.: The value and significance of metagenomics of marine environments, Genomics Proteomics Bioinformatics, 13, 271, available at: https://www.ncbi.nlm.nih.gov/pmc/articles/pmc4678774/ (last access: 25 March 2021), 2015.
Zhou, Z., Meng, H., Liu, Y., Gu, J.-D., and Li, M.: Stratified Bacterial and Archaeal Community in Mangrove and Intertidal Wetland Mudflats Revealed by High Throughput 16S rRNA Gene Sequencing, Front. Microbiol., 8, 2148, https://doi.org/10.3389/fmicb.2017.02148, 2017.
tidal zonationon the prokaryotic sediment communities of a pristine mangrove forest. We observed that the variability in environmental factors between tidal zones results in differences in structure, diversity, and the potential function of prokaryotic populations. This suggests that further work is needed in determining the role tidal microhabitat biodiversity has in mangroves.
tidal zonationon the prokaryotic sediment communities of...