the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 Mar 2022
| 09 Mar 2022
Ideas and perspectives: Sea-level change, anaerobic methane oxidation, and the glacial–interglacial phosphorus cycle
Bjorn Sundby
Pierre Anschutz
Pascal Lecroart
Alfonso Mucci
The oceanic phosphorus cycle describes how phosphorus moves through the ocean, accumulates with the sediments on the seafloor, and participates in biogeochemical reactions. We propose a new two-reservoir scenario of the glacial–interglacial phosphorus cycle. It relies on diagenesis in methane hydrate-bearing sediments to mobilize sedimentary phosphorus and transfer it to the oceanic reservoir during times when falling sea level lowers the hydrostatic pressure on the seafloor and destabilizes methane hydrates. The stock of solid phase phosphorus mobilizable by this process is of the same order of magnitude as the dissolved phosphate inventory of the current oceanic reservoir. The potential additional flux of phosphate during the glacial period is of the same order of magnitude as pre-agricultural, riverine dissolved phosphate fluxes to the ocean. Throughout the cycle, primary production assimilates phosphorus and inorganic carbon into biomass, which, upon settling and burial, returns phosphorus to the sedimentary reservoir. Primary production also lowers the partial pressure of CO2 in the surface ocean, potentially drawing down CO2 from the atmosphere. Concurrent with this slow “biological pump”, but operating in the opposite direction, a “physical pump” brings metabolic CO2-enriched waters from deep-ocean basins to the upper ocean. The two pumps compete, but the direction of the CO2 flux at the air–sea interface depends on the nutrient content of the deep waters. Because of the transfer of reactive phosphorus to the sedimentary reservoir throughout a glaciation cycle, low-phosphorus and high-CO2 deep waters reign at the beginning of a deglaciation, resulting in rapid transfer of CO2 to the atmosphere. The new scenario provides another element to the suite of processes that may have contributed to the rapid glacial–interglacial climate transitions documented in paleo-records.
- Article
(476 KB) - Full-text XML
- BibTeX
- EndNote





Each of the glacial cycles that characterize the Pleistocene lasted roughly 100 kyr. During each cycle, about 90 kyr were glacial (cold), allowing continental ice sheets to build up, and 10 kyr were interglacial (warm), allowing the ice sheets to melt. The large volumes of water transferred from the ocean to the continents during the glacial part of the cycle and the return flow of water to the ocean during the interglacial caused the globally averaged sea level to fall by more than 100 m and then rise again (Siddal et al., 2003; Lambeck et al., 2014). Seasonality, caused by cyclic variations in Earth's orbit around the Sun (Milankovitch cycles), are the fundamental drivers of the glacial cycles, but orbital variations by themselves cannot account for the rapid climate transitions documented in paleo-records. This implies that positive feedbacks within Earth's climate system must amplify the orbital drivers (Sigman and Boyle, 2000). Much research has focused on processes that might provide such feedbacks. Given that carbon dioxide (CO2) is a greenhouse gas and since the ocean is the largest CO2 reservoir on Earth (50 times larger than the atmospheric reservoir), processes that influence the exchange of CO2 between the ocean and the atmosphere top the list of candidates.
Measurements on air samples trapped in ice cores have shown that the partial pressure of CO2 (pCO2) in the atmosphere during the glacial period of a cycle was substantially lower than during the intervening interglacial (Barnola et al., 1987; Petit et al., 1999). The atmospheric pCO2 decreased slowly throughout the glacial part of the cycle, as did sea level. By comparison, both the rise of atmospheric pCO2 and the sea-level rise at the end of the glaciation (termination) were remarkably rapid, lasting 10 kyr or less. The concomitant change in sea level and atmospheric pCO2 through time suggests that the two phenomena are linked, but the nature of the link has remained elusive (Sigman and Boyle, 2000; Kohfeld et al., 2005; Peacock et al., 2006; Sigman et al., 2010; Kölling et al., 2019).
Another greenhouse gas, methane, is abundant in continental margin sediments where it occurs as a solid in the form of methane hydrate (clathrate) (Kvenvolden, 1993; Bohrmann and Torres, 2006; Ruppel and Kessler, 2017). Paull et al. (1991) proposed a direct link between sea-level fall and global warming as follows: as the sea level falls, the pressure on the sediment column decreases, methane hydrates become destabilized, and methane gas is released into the pore water. They further proposed that methane released in this way could reach the atmosphere and initiate a warming event, but it is doubtful that the vast quantities of methane that would be required to trigger substantial warming could reach the atmosphere before being oxidized in the sediments and the overlying water (Archer et al., 2000; Archer, 2007). The search for mechanisms that may explain the glacial–interglacial variation in atmospheric pCO2 has therefore focused on other scenarios.
Common to many scenarios is the idea that the whole ocean inventory of major nutrients may have varied on glacial–interglacial timescales: if the nutrient inventory in the global ocean were to increase, mixing and upwelling could increase the flux of nutrients to the surface ocean. This would stimulate primary productivity, increase the flux of organic matter to the seafloor, and lower the pCO2 in the surface ocean. This scenario is supported by reports that global rate of organic matter burial is maximal during glaciation (Cartapanis et al., 2016; Boyle, 1986; Wallmann, 2010). If the pCO2 in the surface ocean were to fall below the atmospheric pCO2, CO2 would be drawn down from the atmosphere. A temporally variable nutrient inventory could thus explain the glacial–interglacial pCO2 difference. The combination of processes involved in this scenario has become known as the biological pump (e.g., Falkowski, 1997; Hain et al., 2014).
Searching for a temporary sink for phosphorus (P), the nutrient often considered to be limiting on geological timescales in the ocean, Broecker (1982a, b) developed a scenario whereby phosphorus is removed from the global ocean by deposition of P-containing sediment on continental shelves during sea-level high stands and returned to the ocean by erosion of shelf sediments during sea-level low stands. This scenario, known as the “shelf nutrient hypothesis”, suffers a major weakness: the amount of sediment that has to be deposited and eroded on the relevant timescale appears to be far greater than can be supported by observations (Peacock et al., 2006). Another weakness of the hypothesis is the absence of an explicit mechanism that would release phosphate into the aqueous phase once the sediments are eroded. Filippelli et al. (2007) proposed that, with a proper lag time, subaerial weathering by acidic rainfall and plant colonization of exposed shelf sediments will release phosphorus to the ocean. As Archer et al. (2000) stated, it seems that all of the simple mechanisms for lowering pCO2 in the surface ocean have been eliminated. Broecker (2018) made the point that although alternate scenarios may exist to explain glacial cycles, one must take into account that the situation is complex and that no single scenario may be the “best”.
In a recent modelling study, we drew attention to the fact that methane is a fuel that can drive diagenesis in sediments. Using manganese as an example, we showed that methane-fuelled diagenesis can reduce and redistribute solid-phase oxidized manganese throughout the sediment column (Sundby et al., 2015). Building on that study, we now focus on methane-fuelled reduction of oxidized sedimentary iron phases and the consequent release of iron-oxide-bound phosphorus into the pore water. The objective is to develop a chronological scenario of phosphorus cycling in the global ocean over a full glacial cycle. In this scenario, which capitalizes on the two-reservoir model of Broecker (1982a, b), phosphorus cycles between two principal reservoirs: the ocean and the sediments that accumulate on continental margins.
We focus on the mechanisms that promote the release of sedimentary phosphorus into the pore waters and the transfer of phosphorus to the oceanic reservoir. We then explore the mechanisms that return phosphorus to the sedimentary reservoir throughout a glacial cycle. Finally, we examine the interaction of the glacial–interglacial phosphorus cycle with the carbon system to discover how this interaction might affect variations in atmospheric pCO2. The approach focuses on large-scale temporal and spatial processes, but it does not preclude eventual incorporation of shorter timescale and broader spatial-scale processes in a more detailed scenario. By including diagenesis, we can revive elements of Broecker's original hypothesis by providing a set of mechanisms that do not rely on large-scale erosion to release phosphate from sediments. By invoking diagenesis (phosphate released to the pore waters upon the reductive dissolution of iron oxides by sulfide produced during anaerobic methane oxidation), we can show that methane-driven diagenesis may contribute to the redistribution of phosphate between the oceanic and the sedimentary reservoirs.
3.1 Sources and sinks of phosphorus
The inventory of the oceanic phosphorus reservoir depends on the supply of phosphorus from the continents, remineralization of organic phosphorus in the water column and the upper sediment column, burial of solid phases with which phosphorus is associated, release of soluble phosphorus from the sediments, and removal by hydrothermal activity (e.g., Wallmann, 2010). Burial on the abyssal seafloor is the ultimate sink for phosphorus, but an amount equivalent to about half of the total phosphorus flux from the continents to the modern ocean does not reach the abyssal seafloor but settles out and is buried on continental margins (Colman and Holland, 2000; Ruttenberg, 2003). The particulate phosphorus that is delivered to the seafloor contains, on average, one-third each of organic phosphorus, iron-oxide-bound phosphorus, and poorly reactive apatite minerals (Berner et al., 1993; Delaney, 1998; Fillipelli, 1997; Ruttenberg, 2003). Organic phosphorus exists in a variety of forms (primarily phosphate esters) that originate from excretion, decomposition, death, and autolysis of organisms. Microbial degradation of organic matter during the earliest stages of diagenesis converts organic phosphorus to dissolved inorganic phosphate. Part of the remineralized phosphorus escapes into the water column and is added to the oceanic phosphorus reservoir; part of it is co-precipitated with or adsorbed onto iron (hydr)oxide minerals or may be converted to poorly reactive phases such as calcium fluoroapatite (Delaney, 1998).
In its simplest expression, the marine phosphorus cycle consists of a source and a sink of P-bearing material and a set of processes that act upon this material (Fig. 1). Erosion and weathering of continental rocks supply phosphorus to the ocean, and burial on the seafloor removes it. Until recently, the phosphorus sink was thought to be represented by poorly reactive (stable) apatite minerals (Egger et al., 2015). This view presumably had its origin in the analytical procedures that were used to quantify individual P-containing components in marine sediments. This view is now changing, and other phosphorus minerals are thought to be involved. This is exemplified by the authigenesis of vivianite, a hydrous ferrous phosphate that can form in sulfate-poor sediments such as those in brackish estuaries and may act as an important burial sink for phosphorus in brackish environments worldwide (Egger et al., 2015). Vivianite authigenesis requires pore water with elevated ferrous iron (Fe2+) and phosphate concentrations (e.g., Liu et al., 2018). Low-sulfate lacustrine settings typically satisfy these criteria, and the presence of vivianite is commonly reported in freshwater sediments (e.g., Rothe et al., 2016).
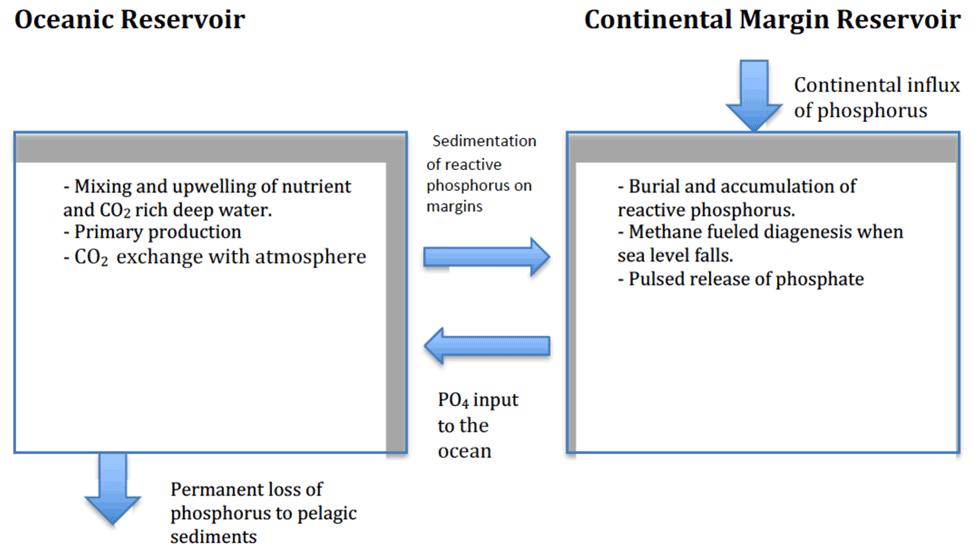
In more sulfate-rich settings, the presence of a sulfate–methane transition zone (SMT) has been shown to provide favourable conditions for vivianite authigenesis (Egger et al., 2015, 2016; Hsu et al., 2014; März et al., 2008, 2018; Slomp et al., 2013). The production of dissolved sulfide by anaerobic oxidation of methane and the associated conversion of Fe oxides to Fe sulfides result in elevated pore-water phosphate concentrations around the SMT (März et al., 2008). The subsequent downward diffusion of phosphate into sulfide-depleted pore water below the SMT may lead to the precipitation of vivianite if sufficient reduced Fe is available (Egger et al., 2015; März et al., 2018).
According to the depositional setting, pore-water phosphate profiles in marine sediments tend to display a concentration maximum below the dissimilatory iron and sulfate reduction zone (e.g., Krom and Berner, 1981), within or near the sulfate–methane transition zone (e.g., März et al., 2008) as well as at greater depths where it is most likely released by deep subsurface organic carbon degradation (e.g., Niewöhner et al., 1998). The associated concentration gradients drive vertical transport of soluble phosphate in both the downward and the upward directions. To sustain these fluxes requires the presence of phosphate sinks both below and above the SMT zone. This conclusion is in agreement with the postulated presence of a phosphate sink and its strength, but it does not provide information about the nature of the sink.
A large body of research shows that phosphate is in fact released from continental margin sediments into the overlying oceanic water. Perhaps the most prominent results of this research are published in the paper by Colman and Holland (2000). They analyzed nearly 200 published pore-water profiles of dissolved phosphate in sediment cores from a variety of marine environments and calculated phosphate fluxes into the overlying water. They concluded that the return flux of phosphate from continental margin sediments was more than 1 order of magnitude larger than the riverine flux of total dissolved phosphorus to the ocean. Hensen et al. (1998) reported phosphate fluxes that are slightly lower but of the same order of magnitude from deep (>1000 m) sediments of the South Atlantic Ocean. Wallmann (2010) used a mass balance approach to estimate phosphate fluxes in the ocean and came up with the remarkable conclusion that the pre-human modern ocean was losing dissolved phosphate (to the sedimentary reservoir) at a rate of about 5 % kyr−1. Studies of phosphorus diagenesis in continental margin sediments have also shown that phosphate is released to the overlying waters (Sundby et al., 1992). These observations point to the important role that diagenesis (oxidation of reduced carbon) plays in the marine phosphorus cycle. Methane being prominently present in continental margin sediments suggests that our scenario, whereby sea-level variations have important consequences for methane oxidation and consequently the marine phosphorus cycle, has merit.
3.2 Phosphorus diagenesis
The term phosphorus usually stands for the sum total of all phosphorus species, organic as well as inorganic, particulate as well as dissolved. It is typically defined operationally according to the analytical methods by which it is determined (e.g., Ruttenberg, 2003). Phosphorus bound to iron oxides is defined operationally as “reactive particulate phosphorus”, but the latter definition typically includes particulate organic phosphorus, phosphate reversibly adsorbed to other mineral surfaces, and phosphate in carbonate fluorapatite minerals and fish bones. In sediment pore waters, soluble reactive phosphate (also referred to as SRP, orthophosphate, or dissolved inorganic phosphorus) is a minor component of the reactive phosphorus pool and occurs chiefly as the hydrogen phosphate species (). Particulate and soluble forms of phosphorus are subjected to different modes of transport. Particulate phosphorus is transported to the seafloor by settling through the oceanic water column and is buried in the sediment. Soluble forms of phosphorus are transported by diffusion along concentration gradients that often develop in sediment pore waters and across the sediment–water interface. Transport by bioturbation and bioirrigation can be important in the upper sediment column.
In this study, we refer to soluble inorganic phosphorus as “phosphate” or “SRP” and to insoluble forms as “particulate phosphorus” or “total phosphorus”. The term reactive iron has been applied to the fraction of iron in marine sediments that reacts readily with sulfide (Canfield, 1989). By analogy, we define reactive particulate phosphorus as the fraction of total P that is adsorbed on or is co-precipitated with iron oxides and can therefore be released to pore waters when iron oxides are reductively dissolved.
The challenge presented by the original shelf nutrient scenario is to find mechanisms that can trigger reciprocal changes in the sedimentary and oceanic phosphorus inventories over a glacial cycle. Diagenesis is a strong candidate for such a mechanism. Diagenesis is defined as the sum of the physical, chemical, and biological processes that bring about changes in a sediment subsequent to deposition (Berner, 1980). Phosphorus diagenesis is intimately linked to the diagenesis of iron oxide minerals, as these are the most important carrier phases for reactive phosphorus in sediments (Ruttenberg, 2003). Surface sediments contain both detrital and authigenic forms of oxidized iron, of which the most reactive fractions are typically poorly crystalline (e.g., ferrihydrite and nano-particulate goethite) and occur in the authigenic fraction. Iron oxides have been characterized operationally by their reactivity towards hydrogen sulfide (Canfield, 1989; Roberts, 2015), and various sequential extraction schemes have been designed and applied to distinguish the reactivity of iron oxides towards a variety of reductants (Anschutz et al., 1998; Kostka and Luther, 1994; Poulton and Canfield, 2005; Ruttenberg, 2003). For a recent review of the extensive literature on sequential extraction procedures, we refer to Anschutz and Deborde (2016).
Authigenic iron oxides form in the upper part of the sediment column above the depth where the stability boundary for the redox couple is located. Reactive iron buried below this boundary is reduced to soluble Fe(II) at a rate that depends on the reactivity of Fe(III) phases and organic matter, the availability of which typically decreases with depth below the sediment surface, as well as sedimentation rate and sulfide exposure time (Canfield, 1989). Fe(II) is then immobilized as sulfides (in organic-rich sediment) or is transported up by diffusion across the redox boundary and reoxidized by various oxidants (O2, , MnO2). This “freshly precipitated” iron oxide is very reactive and can be recycled multiple times across the redox boundary before it is finally permanently buried (Canfield et al., 1993). The presence of authigenic iron oxides in the sediment can be quantified according to its reactivity towards a “weak” reductant such as buffered ascorbic acid (Hyacinthe and Van Cappellen, 2004; Kostka and Luther, 1994). Once buried, authigenic iron oxides may undergo an aging process that includes stepwise dehydration of amorphous phases such as ferrihydrite to goethite and diminishing specific surface area (Lijklema, 1977). This renders reactive iron oxides more refractory, which, in addition to diagenetic remobilization, is reflected by a diminishing content of ascorbate-extractable iron with depth in the sediment (Anschutz et al., 1998). Thus, the bulk reactivity of the sedimentary iron oxides typically decreases with time (depth of burial). The reactivity of the sedimentary organic matter that survives burial below the oxic surface sediment is also important because the reduction rate of sulfate and production rate of H2S depend on it. In the section that follows, we will introduce methane, a highly mobile form of organic matter that can be oxidized anaerobically in sediments by micro-organisms that use sulfate as terminal electron acceptor.
3.3 Linking phosphorus diagenesis to sea-level changes via anaerobic methane oxidation
Much of our understanding of the early stages of diagenesis rests on the notion that diagenesis is fuelled by the organic carbon that settles to the seafloor and is buried and that diagenesis comes to an end when this carbon has been fully consumed. However, in continental margin sediments, where immense sub-surface accumulations of methane are present in the form of methane hydrate (Bohrmann and Torres, 2006; Ruppel and Kessler, 2017), methane can support diagenesis above and beyond that fuelled by organic matter settling from the water column (Burdige and Komada, 2011, 2013; Komada et al., 2016). Methane fuels diagenesis via a number of microbially mediated processes of which the most important is anaerobic microbial oxidation of methane (AOM) using sulfate as the terminal electron acceptor (Boetius et al., 2000; Jørgensen and Kasten, 2006; Nauhaus et al., 2002). AOM has been described as a process that intercepts methane that migrates through anoxic pore water, thereby preventing significant quantities of methane from reaching the atmosphere (e.g., Geprägs et al., 2016). AOM coupled to sulfate reduction does more than just remove methane; it also produces sulfide and bicarbonate (Eq. 1).
The sediment layer within which AOM takes place is known as the sulfate–methane transition zone (SMT). It can be located at depths varying from centimetres to tens of metres below the sediment surface (Borowski et al., 1999). Riedinger et al. (2014) have made the case for coupled anaerobic oxidation of methane via iron oxide reduction, and Egger et al. (2017) have proposed that a potential coupling between iron oxide reduction and methane oxidation likely affects iron cycling and related biogeochemical processes such as burial of phosphorus. Of special interest here is the production of sulfide, a strong reductant that can reductively dissolve even fairly refractory iron oxides and thereby release the associated phosphate into the sediment pore water.
At steady state, the SMT would be located at a fixed distance from the seafloor, and the flux of soluble phosphate from the SMT would be controlled by the burial rate of reactive phosphorus. However, the depth of the SMT in continental margin and epicontinental sediments is not necessarily at steady state on glacial timescales (Henkel et al., 2012). Meister et al. (2007, 2008) linked diagenetic dolomite formation in hemipelagic sediments to sea-level changes, showing that the SMT migrates up and down within the sediment column during the course of 100 kyr cycles. They hypothesized that the SMT persists within an organic-carbon-rich interglacial sediment layer sandwiched between layers of organic-carbon-poor glacial sediment. Contreras et al. (2013) reported evidence that the SMT in organic-carbon-rich sediments from the highly productive Peruvian shelf has migrated vertically in response to cyclic variations in the carbon flux during glacial–interglacial periods. The depth of the SMT may also fluctuate and the upward flux of methane may increase in response to over-pressuring of the underlying gas reservoir, earthquakes, or sediment mass movements (Henkel et al., 2011; Fischer et al., 2013)
The upward displacement and ultimate location of the SMT in the sediment column is critical since the reduction of iron oxides by sulfide occurs at this location and within the displacement interval and releases phosphate to the pore water. The closer the SMT migrates to the sediment surface, the greater the instantaneous flux of sulfate into the sediment and the shorter the path that phosphate travels before it escapes the sediment. The upward displacement of the SMT and the instantaneous flux of phosphate to the ocean can be linked to the rate of sea-level fall if we assume that a time-variable supply of methane from methane-hydrate-bearing sediment layers can also bring about a fluctuating SMT.
3.4 Background (steady-state) fluxes of methane and phosphate in methane-hydrate-bearing sediment
The depth distribution of methane hydrate in sediments is constrained by the relatively narrow pressure and temperature range within which methane hydrate is stable and can exist as a solid (e.g., Ruppel and Kessler, 2017). The lower and upper boundaries of the methane hydrate stability field respond to the accumulation and burial of successive layers of fresh sediment by migrating upward towards the seafloor. Methane hydrate located at the lower stability boundary is thereby transported below this boundary, where it becomes unstable, dissociates, and releases methane into the pore water. Consequently, the downward-directed burial flux of sediment is accompanied by an upward-directed flux of methane.
The lower stability boundary for methane hydrate can be observed seismically as a discontinuity in sound transmission as a gas-rich layer develops. High-resolution seismic data from the Blake Ridge crest (Borowski et al., 1999) show that methane gas, released by hydrate dissociation at the lower stability boundary, can be injected well into the overlying hydrate stability zone. The data indicate that methane, in the form of free gas, can migrate hundreds of metres through the hydrate stability zone before re-forming as solid phase hydrate (Gorman et al., 2002). The ability of gaseous methane to migrate across a thermodynamic regime where it should be trapped as a hydrate suggests that gas migration through the sediment column plays an important role in the interaction of sub-seafloor methane with the overlying ocean. Under steady-state conditions, the upper boundary of the methane hydrate stability field would keep pace with the rate of sediment accumulation, which would create a zone in the upper sediment column within which methane can be removed from the pore water by solid phase hydrate precipitation. In the presence of sulfate, which is in abundant supply in the overlying ocean, methane can also be removed from the pore water by anaerobic methane oxidation. According to the stoichiometry of AOM (Eq. 1), the upward flux of methane into the SMT should equal the downward flux of sulfate. In the absence of electron donors other than methane, a linear pore-water sulfate gradient is expected and can be used to estimate the instantaneous methane flux (Borowski et al., 1999).
3.5 Sea-level changes and methane and phosphate fluxes in the sediment
Because of the sensitivity of methane hydrate deposits to changes in temperature and pressure, one can expect that fluctuations in sea level and temperature, both of which are associated with glacial cycles, will perturb the depth distribution of methane in the sediment. For example, the hydrostatic pressure decrease that was associated with the 120 m sea-level drop during the last glacial maximum has been estimated to lower the hydrostatic pressure enough to raise the lower boundary of the gas hydrate stability field by as much as 20 m (Dillon and Paull, 1983). There can therefore be little doubt that a perturbation such as a sea-level drop can cause methane to be released into the pore water and thereby increase the instantaneous fluxes of methane and phosphate above and beyond the slower background fluxes sustained by steady-state sediment accumulation (Sect. 3.4).
The stoichiometry of anaerobic methane oxidation requires that the flux of sulfate into the sediment increase in order to accommodate an increased upward flux of methane (Eq. 1). This implies that the sulfate gradient grows steeper and that the SMT migrates upward (Fig. 2). As the SMT moves upward, it is accompanied by a front of dissolved sulfide produced by AOM that can reduce buried phosphate-bearing Fe oxides if present. Iron oxide reduction and dissolved ferrous iron production may generate a downward directed flux of Fe(II) into the sediment located below the SMT. It has been suggested (März et al., 2018) that this might be one of the conditions (among others) that would lead to authigenesis of Fe(II) minerals such as vivianite.
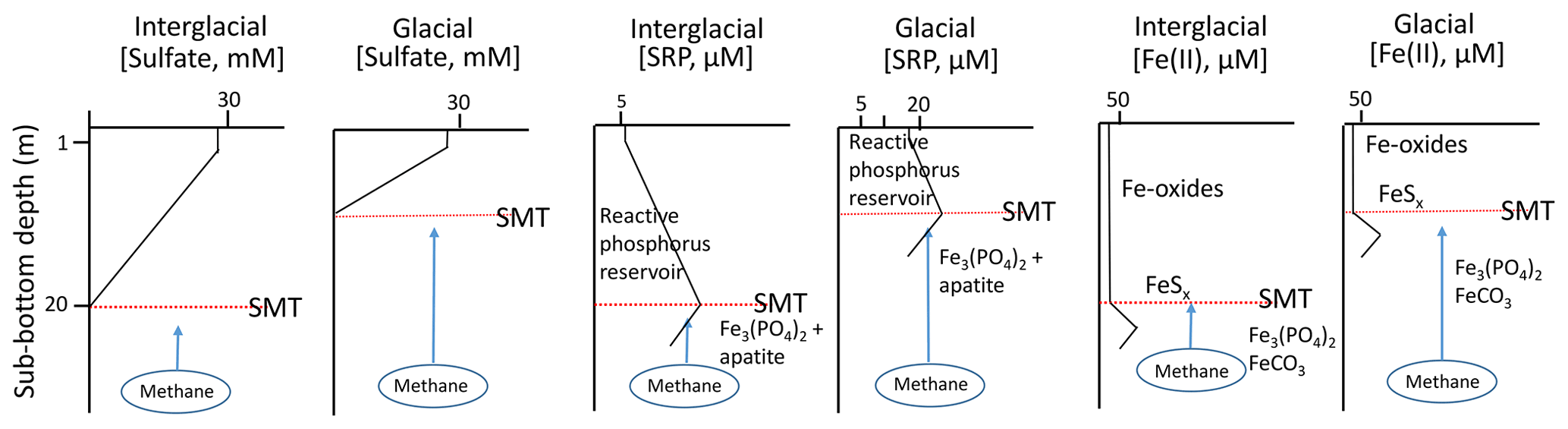
Figure 2Pore-water profiles for relevant species (). Note that the SMT migrates vertically when forced by variable CH4 fluxes. Given the added SRP flux generated by the vertically migrating SMT, the concentration of maximum buffering capacity of the sediment (or zero equilibrium phosphate concentration (EPC0); see Sundby et al. (1992) for details, a concept first introduced by Froelich (1988)),will be rapidly exceeded and the pore-water SRP concentration gradient across the SWI will strengthen. If the flux of SRP from below is large enough, the buffering (or adsorption) capacity of the authigenic iron oxides may be overwhelmed, and the linear gradient could extend all the way to the SWI.
Recent syntheses of global sea-level records (Foster and Rohling, 2013; Lambec et al., 2014; Wallmann et al., 2016) show that there were several episodes of sea-level fall during the last glaciation, each episode interrupted by periods of stable sea level. This suggests that there could also have been several pulses of methane release into the pore water, which would cause the sub-bottom depth of the SMT and the reactions associated with it to fluctuate.
The phosphate flux cannot be expected to directly mirror the fluxes of methane and sulfate because the reductive dissolution rate of the sedimentary iron oxide pool is not constant but depends on the reactivity of the individual iron oxides in the sediment (Canfield, 1989). Furthermore, the efflux of phosphate is ultimately limited by the pool of reactive phosphorus present within the displacement interval of the SMT in the sediment column when sea level falls. If phosphate-bearing iron oxides are absent within the displacement interval (either they never accumulated or were reduced by a previous migration of the SMT) then the phosphate concentration gradient would not be altered significantly, as would the flux of phosphate across the SWI. Even if phosphate-bearing iron oxides are present within the displacement interval, the phosphate gradient and flux across the SWI will wane with time as the phosphate-bearing iron oxides are deactivated (by FeS coating or Fe(II) adsorption) or exhausted (reduced by AOM-generated sulfide). Hence, if the position of the SMT has been invariant for a while, despite a persistent methane flux and/or the presence of methane seeps, we would not expect a significant phosphate flux out of the sediment because phosphate-bearing oxides would either not accumulate in these sediments or have long been dissolved by the sulfidic pore waters, as observed by Niewöhner et al. (1998) in sediments of the upwelling area off Namibia. In fact, pore-water profiles would likely mirror those reported by Wunder et al. (2021) in the Church Trough sediments of South Georgia where cold methane seeps are documented and where little phosphate escapes the sediment. In other words, one would not expect to observe strong phosphate fluxes across the SWI where persistent cold methane seeps are currently found.
3.6 Transport of phosphate in sediment pore waters: adsorption and desorption
Relative to the sediment surface, burial moves reactive particulate phosphorus downward into the sediment column while diffusion moves soluble phosphate upwards. The two oppositely directed phosphorus fluxes interact by partitioning soluble phosphate between solution and sorption sites on solid surfaces, mostly to iron oxides (Krom and Berner, 1980, 1981; Sundby et al., 1992). Phosphate also adsorbs onto other solids, including Mn oxides and carbonate minerals (Millero et al., 2001; Yao and Millero, 1996). It can also form authigenic carbonate fluorapatite and vivianite (März et al., 2018). It has been observed that all the iron oxide surfaces found in oxic continental margin sediments are “saturated” with phosphate (all available sorption sites are occupied) and that the detrital iron oxide fraction is already saturated in phosphate by the time it arrives on the seafloor (Anschutz and Chaillou, 2009). Irrespective, detrital and diagenetic iron oxides in organic-rich sediments have very high but sliding buffering adsorption capacities (Sundby et al., 1992). Hence, if the pore-water phosphate flux to the SWI is increased, much of the phosphate will be intercepted by these oxides, but the concentration of maximum buffering capacity of the sediment (or zero equilibrium phosphate concentration (EPC0), a concept first introduced by Froelich (1998)) will increase and the concentration gradient and flux of phosphate across the SWI will also increase. Sorption should therefore not fully restrict the transport of phosphate diffusing up from the SMT towards and across the sediment–water interface.
Evidence that soluble phosphate can be transported over large depth intervals in sediments without the pore-water profile being visibly affected by sorption onto sediment particles can be found in the dataset of Niewöhner et al. (1998). Four 12–15 m long cores collected in 1300–2000 m water depth from gas-hydrate-bearing sediments on the continental margin off Namibia display linear pore-water phosphate profiles. In two of the cores, the soluble phosphate profile is linear over a 15 m depth interval, from the sediment–water interface to the bottom of the cores; the two other cores display a slope change at about 2 m depth, below which the profile is linear. We do not wish to imply that these profiles are representative of glacial sediments, but they do illustrate an important point: it is possible for soluble phosphate to diffuse over long distances in sediment pore water without encountering significant impediment by secondary diagenetic reactions.
3.7 Quantification of methane-fuelled phosphorus diagenesis
Methane hydrate is thought to exist in the pore spaces of marine sediment located in water depths ranging from 500–4000 m (Paull et al., 1991; Bratton, 1999). This includes sediment located 0–600 m b.s.f. Bratton (1999) estimated that 40 % to 75 % of the total surface area of continental slopes, i.e., 22–41 ×106 km2, contains gas hydrates. The fraction that could be affected by methane hydrate destabilization in response to sea-level fall is located between 500 and 2000 m water depth (Kennett et al., 2003), i.e., 23×106 km2 (Costello et al., 2015).
Destabilization of methane hydrate in the sediment column increases the local rate of sulfate reduction as well as the flux of sulfate into the sulfate–methane transition zone (SMT). The depth interval of sediment over which the SMT initially migrates may contain phosphate-bearing refractory iron oxides that were buried below the zone where catabolic processes are fuelled by the organic carbon delivered to the sediment surface. The concentration of iron-bound P located several decimetres or several metres below the water–sediment interface is on average 2 µmol g−1 (Colman and Holland, 2000; Slomp et al., 1996; Anschutz et al., 1998; Ruttenberg, 2014). Given a mean porosity of 0.6 for silty continental slope sediments buried several metres below the seafloor (e.g., Charbonnier et al., 2019; Schulz and Zabel, 2006), and a bulk dry sediment density of 2.65 (Berner, 1980), the mass of particles in 1 m3 of wet sediment is 0.4 m3×2650 kg m−3 or about 1000 kg. This mass of sediment would contain 2 mol of Fe-bound phosphorus, which means that a 1 m thick sediment pile can potentially release 2 mol of P per square metre or 2 Mmol km−2. When scaled to the surface area of continental slope sediments located between 500 and 2000 m water depth, this represents a standing stock of 46×1012 mol of mobilizable P per metre of sediment thickness.
When the SMT rises during a glacial episode, the maximum thickness of the sediment layer that can release Fe-bound P is equivalent to that corresponding to deposits that accumulated over a 100 000-year glacial cycle. If the vertical migration of the SMT affects more than 100 000 years of sedimentary deposits, a portion of these deposits is then affected by the migrations triggered by two successive glacial cycles. Irrespective, sediment can only release its Fe-bound P once. On the slope, the mean sedimentation rate is estimated at between 15.3 cm kyr−1 (Burwiks et al., 2011) and 42.6 cm kyr−1 (Egger et al., 2018). Therefore, 100 000 years of sediment accumulation corresponds to a sedimentary column that is 15 to 42 m thick, containing between 0.69×1015 and 1.93×1015 mol of Fe-bound phosphorus.
The inventory of dissolved phosphate in the current ocean is estimated at between 2.6×1015 mol and 3×1015 mol (Colman and Holland, 2000; Sarmiento and Gruber, 2006). The inventory of iron-bound P in 100 000 years of sediment accumulation thus represents 23 % to 76 % of the ocean dissolved phosphate inventory. If this inventory is remobilized over the glacial period of 90 000 years, it would correspond to a flux of 0.8 to 2.1×1010 mol yr−1. These values are of the same order of magnitude as pre-agricultural, riverine dissolved phosphate fluxes to the ocean (1.3×1010 mol yr−1; Meybeck, 1982). This is the first time that a quantitative estimate of the magnitude of the sedimentary mobilizable phosphorus reservoir has been made. A comparison of the oceanic P reservoir to the sedimentary reservoir reveals that the two reservoirs are of comparable magnitude. This raises the role of methane-fuelled phosphorus from a somewhat speculative idea to a potentially major player in the glacial–interglacial phosphorus cycle. The prospective flux of phosphate fed by the ascent of the SMT is significant if the diffusional transport of P is sufficient for this phosphate to reach the sediment surface. The flux equation for transport via molecular diffusion is
where J is the phosphate flux across the sediment–water interface, φ is the mean porosity of the sediment, z is the depth coordinate, and () is the phosphate concentration gradient. Ds is the bulk sediment molecular diffusion coefficient, assumed to be equal to ) (Boudreau, 1996), where Do is the diffusion coefficient in water at in situ temperature. Considering a porosity of 0.65 and Do of 0.0124 m2 yr−1 for at 5 ∘C (Schulz and Zabel, 2006), a mean flux of 1×1010 mol yr−1 from a sediment surface of 23×106 km2 ( mol m−2 yr−1) requires a concentration gradient (Δ(SRP)) ) of 0.100 mol m−3 m−1 or 100 µmol L−1 m−1. This gradient is only 2 to 4 times higher than the highest gradient observed on continental slope sediments from long gravity cores by Niewöhner et al. (1998) and Charbonnier et al. (2019), suggesting that molecular diffusion is not an obstacle to the transfer of the inventory of mobilizable phosphate toward the sediment–water interface, as long as the concentration gradient becomes high, as our scenario predicts. A mean diffusive SRP flux of mol m−2 yr−1 is lower than the phosphate return flux fuelled by organic matter regeneration at the sediment–water interface in continental margins (Hensen et al., 1998; Colman and Holland, 2000). However, the increased flux of SRP from below could increase the pore-water concentration gradient immediately below the sediment–water interface (see Sect. 3.6 and Fig. 2 for explanations).
Methane-fuelled P diagenesis thus adds another trigger to the suite of processes that may explain the increased P flux to the ocean during glacial periods, such as the release of P from the continental margin (Filippelli et al., 2007; Filippelli, 2008; Tsandev et al., 2008), the diagenetic P fluxes driven by the early diagenetic microbial degradation of settling organic matter, and the decreased P burial efficiency (Wallman, 2003; Palastanga et al., 2013). The scenario we have developed provides another element to the suite of processes that many have contributed to the rapid glacial–interglacial climate transitions documented in paleo records.
4.1 The concept of a limiting nutrient
A nutrient is limiting if its addition to the system increases the rate of net primary production. Because phosphorus and nitrogen both are essential elements for life, there has been much debate about which of these elements limits photosynthetic primary production in the ocean (Falkowski, 1997; Galbraith et al., 2008; Lenton and Watson, 2000; Smith, 1984; Tyrrell, 1999). Oceanic inventories of nitrogen underwent large changes between glacial and interglacial periods (Ganeshram et al., 1995, 2002). They have been attributed to greatly diminished water column denitrification and consequent increase in the nitrate inventory during glacial periods. In the modern ocean, it appears that nitrogen is the limiting nutrient. Therefore, increasing the flux of phosphate into the ocean would not necessarily increase the rate of primary production. Nevertheless, irrespective of which nutrient element is limiting, primary production assimilates phosphate into biomass, which settles to the seafloor and is buried under successive layers of new sediment. In this way, phosphorus is transferred from the oceanic reservoir to the sedimentary reservoir. Likewise, the net result of methane-fuelled diagenesis is to return phosphate, a nutrient element, from the sedimentary to the oceanic phosphorus reservoir.
4.2 Expansion and contraction of phosphorus reservoirs during a glacial cycle
The central idea of the simple two-reservoir representation of the phosphorus cycle proposed by Broecker (1982a, b) is that the phosphorus inventory in a reservoir can contract and expand on glacial timescales, eventually impacting the CO2 level in the atmosphere. This simple representation of otherwise complex phenomena has stimulated research on the mechanisms that control nutrient fluxes in the ocean.
Boyle (1986) showed that the oceanic phosphorus inventory can vary on a glacial timescale. He concluded, based on the cadmium content of foraminifera and carbon isotope measurements, that the phosphate content of the ocean during the last glacial maximum was 17 % larger than it is at present. Likewise, Wallmann (2010) found that the pre-human modern ocean was losing dissolved phosphate (to the sedimentary P reservoir) at a rate of about 5 % kyr−1. The “lost” phosphate is assimilated into biomass and/or adsorbed onto mineral particles, the settling of which returns reactive phosphorus to the sediment. Colman and Holland (2000), who examined phosphate cycling in modern continental margin sediments, concluded that the current efflux of phosphate from these sediments is about one-half of the total settling flux of particulate reactive phosphorus. The portion that settles through the ocean water column to the seafloor is eventually converted to stable minerals and lost from the oceanic phosphorus cycle.
4.3 Chronology of events during a glacial phosphorus cycle
With the initiation of a glacial cycle, the global temperature decreases, ice builds up on the continents, the sea-level falls, the pressure on the seafloor decreases, and the lower boundary of the methane hydrate stability field in the sediment column shifts upward. Methane hydrate located below the new lower stability boundary becomes unstable and decomposes. Methane gas is released to the pore water, the concentration gradient becomes steeper, and the upward methane flux increases. Upward-migrating methane encounters downward-diffusing sulfate in the sulfate–methane transition zone (SMT). Here, methane is oxidized to CO2 and sulfate is reduced to sulfide (Eq. 1). The reaction between sulfide and ferric minerals, present within the displacement interval of the SMT, reduces Fe(III) to Fe(II), and phosphate associated with these solids is released into the pore waters, increasing the soluble reactive phosphorus concentration gradient and its flux into the oceanic reservoir (Fig. 2) until the ferric minerals are deactivated or exhausted. Irrespective of which nutrient is limiting, primary production in the photic zone assimilates nutrients into biomass, lowering the inventory and concentration of soluble reactive phosphorus and other nutrients in the oceanic reservoir. The fraction of reactive phosphorus that becomes buried on the continental margin is not necessarily lost to the marine phosphorus cycle. It may conceivably become remobilized by diagenesis during an eventual new glacial cycle.
During times when the sea level remains stable, and assuming that sediment accumulation and burial still take place, methane-fuelled diagenesis can nevertheless occur, as the upper and lower boundaries of the methane hydrate stability field track the burial of sediment. Hence, gas hydrates decompose as the lower stability boundary moves up and release methane and subsequently upon AOM at or slightly above the upper stability boundary release phosphate to the pore water. Phosphate is also released during the microbial remineralization of organic matter reaching the seafloor and, hence, a flux of phosphate – what we call a “background” flux – can therefore be delivered to the ocean even when the sea level remains stable. When sea-level change is then superimposed, the net result is to temporarily amplify the phosphate flux to the ocean beyond the background flux.
Sea-level-driven diagenetic transfer of phosphate from sediment to ocean continues throughout the glaciation period, perhaps in spurts because of intervals of stable sea level. When, after the deglaciation, sea level and pressure on the seafloor have reached a new steady state, diagenetically driven fluxes from the sediment stabilize on pre-glaciation background values. Towards the end of the glaciation period, the stratification of the water column destabilizes (e.g., Basak et al., 2018), which facilitates vertical mixing and brings CO2-rich deep water to the photic zone. The biological pump having been weakened by the ongoing biological removal of soluble reactive phosphorus from the oceanic reservoir and the physical pump having gained strength, transport of nutrient-poor andCO2-rich water to the surface ocean will favour the escape (outgassing) of CO2 into the atmosphere. The rise in atmospheric CO2 levels during the glacial period may also have been helped along by the oxidation of iron sulfides in exposed carbonate-rich shelf sediments (Kölling et al., 2019).
Table 1The glacial–interglacial phosphorus cycle: events and consequences.
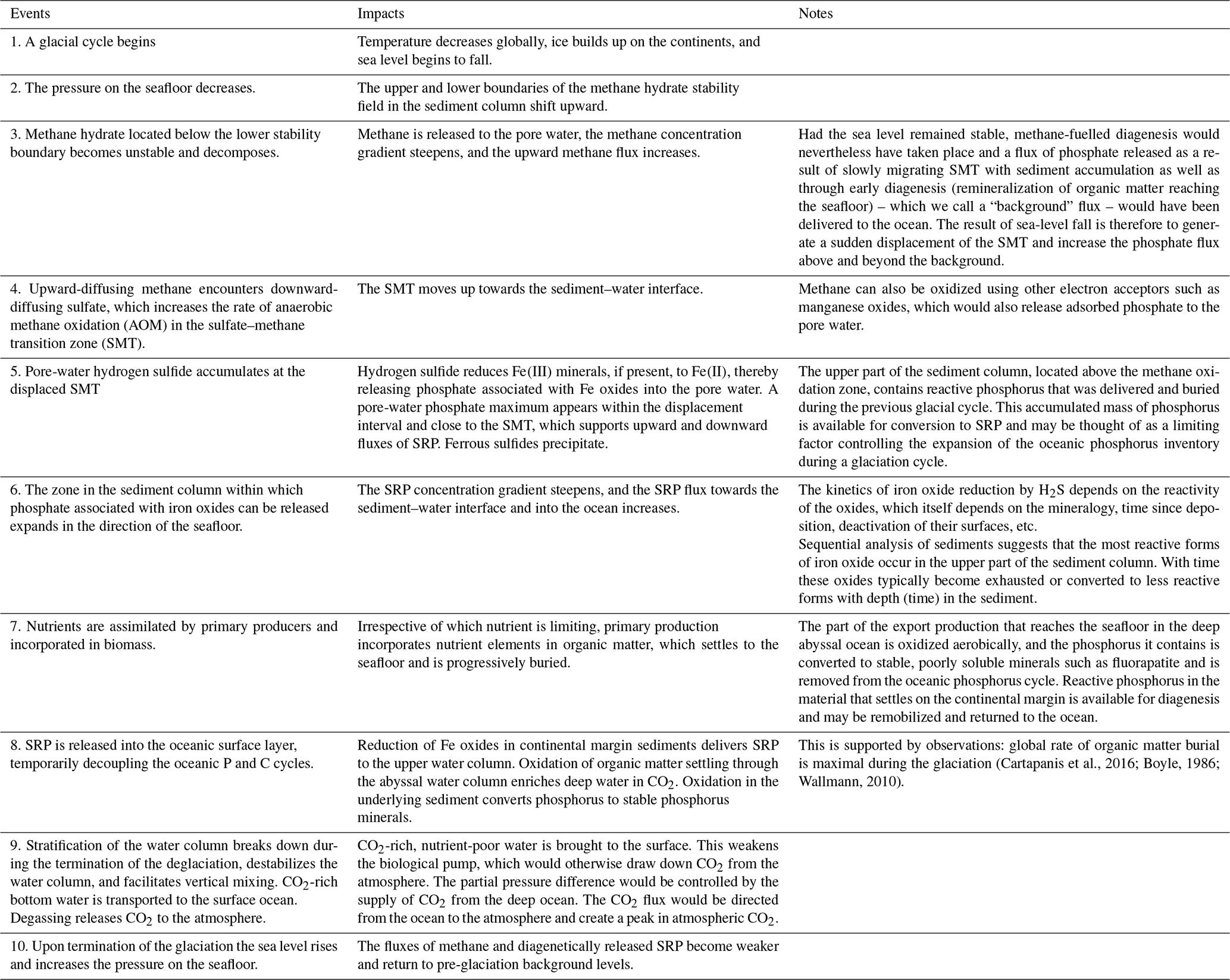
We have modified the two-reservoir scenario of the marine phosphorus cycle proposed by Broecker (1982a, b) to include a diagenetic mechanism that allows for phosphorus to be exchanged between the sedimentary and the oceanic reservoirs on glacial timescales. Within this scenario, a coupled series of processes act upon the glacial–interglacial marine phosphorus cycle (Table 1).
During a glaciation period, water is transferred from the ocean to continental ice sheets. The falling sea level lowers the hydrostatic pressure on the seafloor. The pressure change perturbs the stability field of methane hydrates, and methane gas is released into the pore water. The release of methane gas from gas hydrates amplifies the upward flux of methane through the sediment column. In the sulfate–methane transition zone (SMT), where methane is removed via anaerobic methane oxidation, the increased methane supply increases the demand for sulfate. The sulfate gradient steepens, and the SMT zone moves closer to the sediment–water interface. Reactions within the SMT produce hydrogen sulfide, which reductively dissolves iron oxides within the displacement interval and near the SMT. Iron-bound phosphate can then be released into the pore water. The upward-directed phosphate gradient steepens, which increases the flux of phosphate towards and across the sediment–water interface and increases the phosphate inventory of the ocean.
Phosphorus is transferred from the oceanic to the sedimentary reservoir by sedimentation and burial of phosphorous-containing biogenic particulate matter resulting from primary production and abiotic particulate matter on which phosphate is adsorbed. Unlike the release of phosphate from the sediment, the return flux of phosphorus to the sediment and the associated depletion of the oceanic reactive phosphorus inventory do not occur as a single event but take place throughout the glacial cycle. Burial of organic matter causes a corresponding loss of phosphorus from the oceanic inventory. If phosphorus were the nutrient limiting primary production at the time leading up to the release, primary production would be stimulated by the added phosphate to the surface ocean, CO2 would be drawn down from the atmosphere, and the phosphorus inventory would begin to decrease. If, on the other hand, nitrogen nutrients were to become limiting, which is likely when new phosphate is added to the surface ocean reservoir, the rates of primary production, phosphorus burial, and draw-down of atmospheric CO2 would slow down until the fixed nitrogen-to-phosphorus ratio became similar to the Redfield ratio. It is therefore not unreasonable to expect that with a gradual depletion of phosphorus in the ocean, a stable N:P ratio would develop, co-limiting primary production in the ocean (Lenton and Watson, 2000; Lenton and Klausmeier, 2007). As the glaciation progresses, the ocean will gradually become nutrient depleted irrespective of which is the limiting nutrient. This would weaken the biological pump relative to the physical pump (upwelling of deep CO2-rich water) and set the stage for a tipping point beyond which atmospheric CO2 is controlled by upwelling of CO2-rich deep water. The next glaciation period, which is accompanied by sea-level fall, methane-hydrate decomposition, anaerobic methane oxidation, and phosphate release, allows the biological pump to once again take control over atmospheric CO2.
The proposed scenario capitalizes on elements of Broecker's model and provides another potential trigger to the suite of processes that may have contributed to the rapid glacial–interglacial climate transitions documented in paleo-records.
The central idea of the simple two-reservoir representation of the phosphorus cycle proposed by Broecker (1982a, b) is that the phosphorus inventory in a reservoir can contract and expand on glacial timescales, eventually impacting the CO2 level in the atmosphere. This simple representation of otherwise complex phenomena has stimulated research on the mechanisms that control nutrient fluxes in the ocean.
No new data are presented in this paper, which is inspired by and developed with data in the public domain. These are cited in the references included in the paper.
BS, PA, PL, and AM designed the study. BS wrote the manuscript with input from all authors.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Bjorn Sundby and Alfonso Mucci gratefully acknowledge the financial support from the Natural Sciences and Engineering Research Council of Canada through their Discovery grants. We wish to thank the three journal reviewers, David Archer, Gabriel Filippelli, and Sabine Kasten, for their incisive comments on a previous version of this paper.
This research has been supported by the NSERC of Canada (grant no. RGPIN/04421–2018).
This paper was edited by Tina Treude and reviewed by Gabriel Filippelli, Sabine Kasten, and David Archer.
Anschutz, P. and Chaillou, G.: Deposition and fate of reactive Fe, Mn, P, and C in suspended particulate matter in the Bay of Biscay, Cont. Shelf Res., 29, 1038–1043, 2009.
Anschutz, P. and Deborde, J.: Spectrophotometric determination of phosphate in matrices from sequential leaching of sediments, Limnol. Oceanogr.-Meth., 14, 245–256, 2016.
Anschutz, P., Zhong, S., Sundby, B., Mucci, A., and Gobeil, C.: Burial efficiency of phosphorus and the geochemistry of iron in continental margin sediments, Limnol Oceanogr., 43, 53–64, 1998.
Archer, D.: Methane hydrate stability and anthropogenic climate change, Biogeosciences, 4, 521–544, https://doi.org/10.5194/bg-4-521-2007, 2007.
Archer, D., Winguth, A., Lea, D., and Mahowald, N.: What caused the glacial/interglacial atmospheric pCO2 cycles?, Rev. Geophys., 38, 159–189, 2000.
Barnola, J. M., Raynaud, D., Korotkevich, Y. S., and Lorius, C.: Vostok ice core provides 160 000-year record of atmospheric CO2, Nature, 329, 408–414, 1987.
Basak, C., Fröllje, H., Lamy, F., Gersonde, R., Benz, V., Anderson, R. F., Molina-Kercher, M., and Pahnke, K.: Breakup of last glacial deep stratification in the South Pacific, Science, 359, 900–904, 2018.
Berner, R. A.: Early Diagenesis: A Theoretical Approach, Princeton University Press, ISBN 9780691082608, 1980.
Berner, R. A., Ruttenberg, K. C., Ingall, E. D., and Rao, J. L.: The nature of phosphorus burial in modern marine sediments, in: Interactions of C, N, P and S biogeochemical cycles and global change, edited by: Wollast, R., Mackenzie, F. T., and Chou, L., Springer, Berlin, Heidelberg, 365–378, ISBN 978-3-642-76066-2, 1993.
Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gieseke, A., Amann, R., Jørgensen, B. B., Witte, U., and Pfannkuche, O.: A marine microbial consortium apparently mediating anaerobic oxidation of methane, Nature, 407, 623–626, 2000.
Bohrmann, G. and Torres, M. E.: Gas hydrates in marine sediments, in: Marine Geochemistry, edited by: Schulz, H. D. and Zabel, M., Springer, Berlin, Heidelberg, 481–512, ISBN 978-3-540-32143-9, 2006.
Borowski, W. S., Paull, C. K., and Ussler, W.: Global and local variations of interstitial sulfate gradients in deep-water, continental margin sediments: Sensitivity to underlying methane and gas hydrates, Mar. Geol., 159, 131–154, 1999.
Boudreau, B. P.: The diffusive tortuosity of fine-grained unlithified sediments, Geochim. Cosmochim. Ac., 60, 3139–3142, 1996.
Boyle, E. A.: Paired carbon isotope and cadmium data from benthic foraminifera: Implications for changes in oceanic phosphorus, oceanic circulation, and atmospheric carbon dioxide, Geochim. Cosmochim. Ac., 50, 265–276, 1986.
Bratton, J. F.: Clathrate eustacy: Methane hydrate melting as a mechanism for geologically rapid sea-level fall, Geology, 27, 915–918, 1999.
Broecker, W. S: Glacial to interglacial changes in ocean chemistry, Prog. Oceanogr., 11, 151–197, 1982a.
Broecker, W. S.: Ocean chemistry during glacial time, Geochim. Cosmochim. Ac., 46, 1689–1705, 1982b.
Broecker, W. S.: Glacial Cycles, Geochem. Perspect., 7, 167–181, 2018.
Burdige, D. J. and Komada, T.: Anaerobic oxidation of methane and the stoichiometry of remineralization processes in continental margin sediments, Limnol. Oceanogr., 56, 1781–1796, 2011.
Burdige, D. J. and Komada, T.: Using ammonium pore water profiles to assess stoichiometry of deep remineralization processes in methanogenic continental margin sediments, Geochem. Geophy. Geosy., 14, 1626–1643, 2013.
Burwicz, E. B., Rüpke, L. H., and Wallmann, K.: Estimation of the global amount of submarine gas hydrates formed via microbial methane formation based on numerical reaction-transport modeling and a novel parameterization of Holocene sedimentation, Geochim. Cosmochim. Ac., 75, 4562–4576, 2011.
Canfield, D. E.: Reactive iron in marine sediments, Geochim. Cosmochim. Ac., 53, 619–632, 1989.
Canfield, D. E., Thamdrup, B., and Hansen, J. W.: The anaerobic degradation of organic matter in Danish coastal sediments: iron reduction, manganese reduction, and sulfate reduction, Geochim. Cosmochim. Ac., 57, 3867–3883, 1993.
Cartapanis, O., Bianchi, D., Jaccard, S. L., and Galbraith, E. D.: Global pulses of organic carbon burial in deep-sea sediments during glacial maxima, Nat. Comm., 7, 1–7, 2016.
Charbonnier, C., Mouret, A., Howa, H., Schmidt, S., Gillet, H., and Anschutz, P.: Quantification of diagenetic transformation of continental margin sediments at the Holocene time scale, Cont. Shelf Res., 180, 63–74, 2019.
Colman, A. S. and Holland, H. D.: The global diagenetic flux of phosphorus from marine sediments to the oceans: redox sensitivity and the control of atmospheric oxygen levels, SEPM Spec. P., 66, 53–75, https://doi.org/10.2110/pec.00.66.0053, 2000.
Contreras, S., Meister, P., Liu, B., Prieto-Mollar, X., Hinrichs, K. U., Khalili, A., Felderman, T. G., Kuypers, M. M. M., and Jørgensen, B. B.: Cyclic 100-ka (glacial-interglacial) migration of subseafloor redox zonation on the Peruvian shelf, P. Natl. Acad. Sci. USA, 110, 18098–18103, 2013.
Costello, M. J., Smith, M., and Fraczek, W.: Correction to Surface area and the seabed area, volume, depth, slope, and topographic variation for the world's seas, oceans, and countries, Environ. Sci. Tech., 49, 7071–7072, 2015.
Delaney, M.: Phosphorus accumulation in marine sediments and the oceanic phosphorus cycle, Global Biogeochem. Cy., 12, 563–572, 1998.
Dillon, W. P., and Paull, C. K.: Marine gas-hydrate II: Geophysical evidence, in: Natural Gas Hydrates., Properties, Occurrence and Recovery, edited by: Cox, J. L., Butterworth, Boston, Mass., 73–90, ISBN 978-0250406319, 1983.
Egger, M., Jilbert, T., Behrends, T., Rivard, C., and Slomp, C. P.: Vivianite is a major sink for phosphorus in methanogenic coastal surface sediments, Geochim. Cosmochim. Ac., 169, 217–235, 2015.
Egger, M., Kraal, P., Jilbert, T., Sulu-Gambari, F., Sapart, C. J., Röckmann, T., and Slomp, C. P.: Anaerobic oxidation of methane alters sediment records of sulfur, iron and phosphorus in the Black Sea, Biogeosciences, 13, 5333–5355, https://doi.org/10.5194/bg-13-5333-2016, 2016.
Egger, M., Hagens, M., Sapart, C. J., Dijkstra, N., van Helmond, N. A. G. M., Mogollón, J. M., Risgaard-Peterson, N., van der Veen, C., Kasten, S., Riedenger, N., Böttcher, M. E., Röchmann, T., Jørgensen, B. B., and Slomp, C. P.: Iron oxide reduction in methane-rich deep Baltic Sea sediments, Geochim.. Cosmochim. Ac., 207, 256–276, 2017.
Egger, M., Riedinger, N., Mogollón, J. M., and Jørgensen, B. B.: Global diffusive fluxes of methane in marine sediments, Nat. Geosci., 11, 421–425, 2018.
Falkowski, P. G.: Evolution of the nitrogen cycle and its influence on the biological sequestration of CO2 in the ocean, Nature, 387, 272–275, 1997.
Filippelli, G. M.: Controls on phosphorus concentration and accumulation in oceanic sediments, Mar. Geol., 139, 231–240, 1997.
Filippelli, G. M.: The global phosphorus cycle: past, present, and future, Elements, 4, 89–95, 2008.
Filippelli, G. M., Latimer, J. C., Murray, R. W., and Flores, J. A.: Productivity records from the Southern Ocean and the equatorial Pacific Ocean: Testing the Glacial Shelf-Nutrient Hypothesis, Deep-Sea Res. Pt. II, 54, 2443–2452, 2007.
Fischer, D., Mogollón, J. M., Strasser, M., Pape, T., Bohrmann, G., Fekete, N., Spiess, V., and Kasten, S.: Subduction zone earthquake as potential trigger of submarine hydrocarbon seepage, Nature Geosci., 6, 647–651, 2013.
Foster, G. L. and Rohling, E. J.: Relationship between sea-level and climate forcing by CO2 on geological timescales, P. Natl. Acad. Sci. USA, 110, 1209–1214, 2013.
Froelich, P. N.: Kinetic control of dissolved phosphate in natural rivers and estuaries: A primer on the phosphate buffer mechanism, Limnol. Oceanogr., 33, 649–668, 1988.
Galbraith, E. D, Sigman, D., Pedersen, T., and Robinson, R. S.: Nitrogen in past marine environments, in: Nitrogen in the Marine Environment, edited by: Capone, D., Bronk, M., Mulholland, M., and Carpenter, D., 2nd edn., Elsevier, 1497–1535, https://doi.org/10.1016/B978-0-12-372522-6.00034-7, 2008.
Ganeshram, R. S., Pedersen, T. F., Calvert, S. E., and Murray, J. W.: Large changes in oceanic nutrient inventories from glacial to interglacial periods, Nature, 376, 755–758, 1995.
Ganeshram, R. S., Pedersen, T. F., Calvert, S., and François, R.: Reduced nitrogen fixation in the glacial ocean inferred from changes in marine nitrogen and phosphorus inventories, Nature, 415, 156–159, 2002.
Geprägs, P., Torres, M. A., Mau, S., Kasten, S., Römer, M., and Bohrmann, G.: Carbon cycling fed by methane seepage at the shallow Cumberland Bay, South Georgia, sub-Antarctic, Geochem. Geophy. Geosy., 17, 1401–1418, 2016.
Gorman, A. R., Holbrook, W. S., Hornbach, M. J., Hackwith, K. L., Lizarralde, D., and Pecher, I.: Migration of methane gas through the hydrate stability zone in a low-flux hydrate province, Geology, 30, 327–330, 2002.
Hain, M. P., Sigman, D. M., and Haug, G. H.: The Biological Pump in the Past. Treatise on Geochemistry (2nd edn.), The Oceans and Marine Geochemistry, 8, 485–517, 2014.
Henkel, S., Strasser, M., Schwenk, T., Hanebuth, T. J. J., Hüsener, J., Arnold, G. L., Winkelmann, D., Formolo, M., Tomasini, J., Krastel, S., and Kasten, S.: An interdisciplinary investigation of a recent submarine mass transport deposit at the continental margin off Uruguay, Geochem. Geophy. Geosy., 12, 1–19, 2011.
Henkel, S., Mongollòn, J. M., Nöthen, K., Franke, C., Bogus, K., Robin, E., Bahr, A., Blumenberg, M., Pape, T., Siefert, R., März, C., de Lange, G. J., and Kasten, S.: Diagenetic barium cycling in Black Sea sediments – A case study for anoxic marine environments, Geochim. Cosmochim. Ac., 88, 88–105, 2012.
Hensen, C., Landenberger, H., Zabel, M., and Schulz, H. D.: Quantification of diffusive benthic fluxes of nitrate, phosphate, and silicate in the southern Atlantic Ocean, Global Biogeochem. Cy., 12, 193–210, 1998.
Hsu, T. W., Jiang, W. T., and Wang, Y.: Authigenesis of vivianite as influenced by methane-induced sulfidization in cold-seep sediments off southwestern Taiwan, J. Asian Earth Sci., 89, 88–97, 2014.
Hyacinthe, C. and Van Cappellen, P.: An authigenic iron phosphate phase in estuarine sediments: composition, formation and chemical reactivity, Mar. Chem., 91, 227–251, 2004.
Jørgensen, B. B. and Kasten, S.: Sulfur cycling and methane oxidation, in: Marine Geochemistry, edited by: Schulz, H. D. and Zabel, M., Springer, Berlin, Heidelberg, 271–302, ISBN 978-3-540-32143-9, 2006.
Kennett, J. P., Cannariato, K. G., Hendy, I. L., and Behl, R. J.: Methane hydrates in Quaternary climate change: The clathrate gun hypothesis, American Geophysical Union, Washington, DC, ISBN 0-87590-296-0, 2003.
Kohfeld, K. E., Le Quéré, C., Harrison, S. P., and Anderson, R. F.: Role of marine biology in glacial-interglacial CO2 cycles, Science, 308, 74–78, 2005.
Kölling, M, Bouimetarhan, I., Bowles, M. W., Felis, T, Goldhammer, T, Hinrichs, K.-U., Schulz, M., and Zabel, M: Consistent CO2 release by pyrite oxidation on continental shelves prior to glacial terminations, Nat. Geosci., 12, 929–934, 2019.
Komada, T., Burdige, D. J., Li, H. L., Magen, C., Chanton, J. P., and Cada, A. K.: Organic matter cycling across the sulfate-methane transition zone of the Santa Barbara Basin, California Borderland, Geochim. Cosmochim. Ac., 176, 259–278, 2016.
Kostka, J. E. and Luther III, G. W.: Partitioning and speciation of solid phase iron in saltmarsh sediments, Geochim. Cosmochim. Ac., 58, 1701–1710, 1994.
Krom, M. D. and Berner, R. A.: The diffusion coefficients of sulfate, ammonium, and phosphate ions in anoxic marine sediments, Limnol. Oceanogr., 25, 327–337, 1980.
Krom, M. D. and Berner, R. A.: The diagenesis of phosphorus in a nearshore marine sediment, Geochim. Cosmochim, Ac., 45, 207–216, 1981.
Kvenvolden, K. A.: Gas hydrates – Geological perspective and global change, Rev. Geophys., 31, 173–187, 1993.
Lambeck, K., Rouby, H., Purcell, A., Sun, Y., and Sambridge, M.: Sea-level and global ice volumes from the Last Glacial Maximum to the Holocene, P. Natl. Acad. Sci. USA, 111, 15296–15303, 2014.
Lenton, T. M. and Klausmeier, C. A.: Biotic stoichiometric controls on the deep ocean N:P ratio, Biogeosciences, 4, 353–367, https://doi.org/10.5194/bg-4-353-2007, 2007.
Lenton, T. M. and Watson, A. J.: Redfield revisited: 1. Regulation of nitrate, phosphate, and oxygen in the ocean, Global Biogeochem. Cy., 14, 225–248, 2000.
Lijklema, L.: The role of iron in the exchange of phosphate between water and sediments, in: Interactions Between Sediments and Fresh Water, edited by: Golterman, H. L., Dr. W. Junk Publishers, The Hague, 313–317, ISBN 9789022006320, 1977.
Liu, J., Izon, G., Wang, J., Antler, G., Wang, Z., Zhao, J., and Egger, M.: Vivianite formation in methane-rich deep-sea sediments from the South China Sea, Biogeosciences, 15, 6329–6348, https://doi.org/10.5194/bg-15-6329-2018, 2018.
März, C., Hoffmann, J., Bleil, U., de Lange, G., and Kasten, S.: Diagenetic changes of magnetic and geochemical signals by anaerobic methane oxidation in sediments of the Zambezi deep-sea fan (SW Indian Ocean), Mar. Geol., 255, 118–130, 2008.
März, C., Riedinger, N., Sena, C., and Kasten, S.: Phosphorus dynamics around the sulphate-methane transition in continental margin sediments: Authigenic apatite and Fe(II) phosphates, Mar. Geol., 404, 84–96, 2018.
Meister, P., McKenzie, J. A., Vasconcelos, C., Bernasconi, S., Frank, M., Gutjahr, M., and Schrag, D. P.: Dolomite formation in the dynamic deep biosphere: results from the Peru Margin, Sedimentology, 54, 1007–1032, 2007.
Meister, P., Bernasconi, S. M., Vasconcelos, C., and McKenzie, J. A.: Sea-level changes control diagenetic dolomite formation in hemipelagic sediments of the Peru Margin, Mar. Geol., 252, 166–173, 2008.
Meybeck, M.: Carbon, nitrogen, and phosphorus transport by world rivers, Am. J. Sci., 282, 401–450, 1982.
Millero, F. J., Huang, F., Zhu X., and Zhang, J.-Z.: Adsorption and desorption of phosphate on calcite and aragonite in seawater, Aquat. Geochem., 7, 33–56, 2001.
Nauhaus, K., Boetius, A., Krüger, M., and Widdel, F.: In vitro demonstration of anaerobic oxidation of methane coupled to sulphate reduction in sediment from a marine gas hydrate area, Environ. Microbiol., 4, 296–305, 2002.
Niewöhner, C., Hensen, C., Kasten, S., Zabel, M., and Schulz, H. D.: Deep sulfate reduction completely mediated by anaerobic methane oxidation in sediments of the upwelling area off Namibia, Geochim. Cosmochim. Ac., 62, 455–464, 1998.
Palastanga, V., Slomp, C. P., and Heinze, C.: Glacial-interglacial variability in ocean oxygen and phosphorus in a global biogeochemical model, Biogeosciences, 10, 945–958, https://doi.org/10.5194/bg-10-945-2013, 2013.
Paull, C. K., Ussler, W., and Dillon, W. P.: Is the extent of glaciation limited by marine gas hydrates?, Geophys. Res. Lett., 18, 432–434, 1991.
Peacock, S., Lane, E., and Restrepo, J. M.: A possible sequence of events for the generalized glacial-interglacial cycle, Global Biogeochem. Cy., 20, GB2010, doi.org/10.1029/2005GB002448, 2006.
Petit, J. R., Jouzel, J., Raynaud, D., Barkov, N. I., Barnola, J. M., Basile, I., Bender, M., Chappellaz, J., Davis, M., Delaygue, G., Delmotte, M., Kotlyakov, V. M., Legrand, M., Lipenkov, V. Y., Lorius, C., Pépin, L., Ritz, C., Saltzman, E., and Stievenard, M.: Climate and atmospheric history of the past 420 000 years from the Vostok ice core, Antarctica, Nature, 399, 429–436, 1999.
Poulton, S. W. and Canfield, D. E.: Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates, Chem. Geol., 214, 209–221, 2005.
Riedinger, N., Formolo, M. J., Lyons, T. W., Henkel, S., Beck, A., and Kasten, S.: An inorganic geochemical argument for coupled anaerobic oxidation of methane and iron reduction in marine sediments, Geobiology, 12, 172–181, 2014.
Roberts, A. P.: Magnetic mineral diagenesis, Earth-Science Rev., 151, 1–47, 2015.
Rothe, M., Kleeberg, A., and Hupfer, M.: The occurrence, identification and environmental relevance of vivianite in waterlogged soils and aquatic sediments, Earth-Sci. Rev., 158, 51–64, 2016.
Ruppel, C. D. and Kessler, J. D.: The interaction of climate change and methane hydrates, Rev. Geophys., 55, 126–168, 2017.
Ruttenberg, K. C.: The global phosphorus cycle, in: Treatise on Geochemistry, edited by: Schleisinger, W. H., Elsevier, 585–643, ISBN 0-08-044343-5, 2003.
Ruttenberg, K. C.: The global phosphorus cycle, in: Treatise on Geochemistry, 2nd edn., edited by: Karl, D. M. and Schlesinger, W. H., Elsevier Science, 499–558, https://doi.org/10.1016/B978-0-08-095975-7.00813-5, 2014.
Sarmiento, J. J. and Gruber, N.: Ocean Biogeochemical Dynamics, Princeton University Press, Princeton, ISBN 9780691017075, 2006.
Schulz, H. D. and Zabel, M.: Marine Geochemistry, 2nd edn., Springer-Verlag, Berlin, Heidelberg, ISBN 978-3-540-32143-9, 2006.
Siddall, M., Rohling, E. J., Almogi-Labin, A., Hemleben, C., Meischner, D., Schmelzer, I., and Smeed, D. A.: Sea-level fluctuations during the last glacial cycle, Nature, 423, 853–858, 2003.
Sigman, D. M. and Boyle, E. A.: Glacial/interglacial variations in atmospheric carbon dioxide. Nature, 407, 859–869, 2000.
Sigman, D. M., Hain, M. P., and Haug, G. H.: The polar ocean and glacial cycles in atmospheric CO2 concentration, Nature, 466, 47–55, 2010.
Slomp, C. P., Epping, E. H. G., Helder, W., and Van Raaphorst, W.: A key role for iron-bound phosphorus in authigenic apatite formation in North Atlantic continental platform sediments, J. Mar. Res., 54, 1179–1205, 1996.
Slomp, C. P., Mort, H. P., Jilbert, T., Reed, D. C., Gustafsson, B. G., and Wolthers, M.: Coupled dynamics of iron and phosphorus in sediments of an oligotrophic coastal basin and the impact of anaerobic oxidation of methane, PloS ONE, 8, e62386, https://doi.org/10.1371/journal.pone.0062386, 2013
Smith, S. V.: Phosphorus versus nitrogen limitation in the marine environment, Limnol. Oceanogr., 29, 1149–1160, 1984.
Sundby, B., Gobeil, C., Silverberg, N., and Mucci A.: The phosphorus cycle in coastal marine sediments, Limnol. Oceanogr., 37, 1129–1145, 1992.
Sundby, B., Lecroart, P., Anschutz, P., Katsev, S., and Mucci, A.: When deep diagenesis in Arctic Ocean sediments compromises manganese-based geochronology, Mar. Geol., 366, 62–68, 2015.
Tsandev, I., Slomp, C. P., and Van Cappellen, P.: Glacial-interglacial variations in marine phosphorus cycling: Implications for ocean productivity, Global Biogeochem. Cy., 22, GB4004, https://doi.org/10.1029/2007GB003054, 2008.
Tyrrell, T.: The relative influences of nitrogen and phosphorus on oceanic primary production, Nature, 400, 525–531, 1999.
Wallmann, K.: Feedbacks between oceanic redox states and marine productivity: A model perspective focused on benthic phosphorus cycling, Global Biogeochem. Cy., 17, 1084, https://doi.org/10.1029/2002GB001968, 2003.
Wallmann, K.: Phosphorus imbalance in the global ocean?, Global Biogeochem. Cy., 24, GB4030, https://doi.org/10.1029/2009GB003643, 2010.
Wallmann, K., Schneider, B., and Sarnthein, M.: Effects of eustatic sea-level change, ocean dynamics, and nutrient utilization on atmospheric pCO2 and seawater composition over the last 130 000 years: a model study, Clim. Past, 12, 339–375, https://doi.org/10.5194/cp-12-339-2016, 2016.
Wunder, L. C., Aromokeye, D. A., Yin, X., Richter-Heitmann, T., Willis-Poratti, G., Schnakenberg, A., Otersen, C., Dohrmann, I., Römer, M., Bohrmann, G., Kasten, S., and Friedrich, M. W.: Iron and sulfate reduction structure microbial communities in (sub-)Antarctic sediments, ISME J., 15, 3587–3604, 2021.
Yao, W. and Millero, F. J.: Adsorption of phosphate on manganese dioxide in seawater, Environ. Sci. Tech., 30, 536–541, 1996.
- Abstract
- Introduction
- Objectives and approach
- Diagenesis and fluxes of phosphorus in continental margin sediments
- Linking the phosphorus cycle to changes in atmospheric CO2
- Summary and conclusions
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Abstract
- Introduction
- Objectives and approach
- Diagenesis and fluxes of phosphorus in continental margin sediments
- Linking the phosphorus cycle to changes in atmospheric CO2
- Summary and conclusions
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References