the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Jan 2020
| 16 Jan 2020
A meta-analysis of microcosm experiments shows that dimethyl sulfide (DMS) production in polar waters is insensitive to ocean acidification
Frances E. Hopkins
Philip D. Nightingale
John A. Stephens
C. Mark Moore
Sophie Richier
Gemma L. Cripps
Stephen D. Archer
Emissions of dimethylsulfide (DMS) from the polar oceans play a key role in atmospheric processes and climate. Therefore, it is important to increase our understanding of how DMS production in these regions may respond to climate change. The polar oceans are particularly vulnerable to ocean acidification (OA). However, our understanding of the polar DMS response is limited to two studies conducted in Arctic waters, where in both cases DMS concentrations decreased with increasing acidity. Here, we report on our findings from seven summertime shipboard microcosm experiments undertaken in a variety of locations in the Arctic Ocean and Southern Ocean. These experiments reveal no significant effects of short-term OA on the net production of DMS by planktonic communities. This is in contrast to similar experiments from temperate north-western European shelf waters where surface ocean communities responded to OA with significant increases in dissolved DMS concentrations. A meta-analysis of the findings from both temperate and polar waters (n=18 experiments) reveals clear regional differences in the DMS response to OA. Based on our findings, we hypothesize that the differences in DMS response between temperate and polar waters reflect the natural variability in carbonate chemistry to which the respective communities of each region may already be adapted. If so, future temperate oceans could be more sensitive to OA, resulting in an increase in DMS emissions to the atmosphere, whilst perhaps surprisingly DMS emissions from the polar oceans may remain relatively unchanged. By demonstrating that DMS emissions from geographically distinct regions may vary in their response to OA, our results may facilitate a better understanding of Earth's future climate. Our study suggests that the way in which processes that generate DMS respond to OA may be regionally distinct, and this should be taken into account in predicting future DMS emissions and their influence on Earth's climate.
- Article
(3080 KB) - Full-text XML
-
Supplement
(1503 KB) - BibTeX
- EndNote




The trace gas dimethylsulfide (DMS) is a key ingredient in a cocktail of gases that exchange between the ocean and atmosphere. Dissolved DMS is produced via the enzymatic breakdown of dimethylsulfoniopropionate (DMSP), a secondary algal metabolite implicated in a number of cellular roles, including the regulation of carbon and sulfur metabolism via an overflow mechanism (Stefels, 2000) and protection against oxidative stress (Sunda et al., 2002). Oceanic DMS emissions amount to 17–34 Tg S yr−1, representing 80 %–90 % of all marine biogenic S emissions and up to 50 % of global biogenic emissions (Lana et al., 2011). DMS and its oxidation products play vital roles in atmospheric chemistry and climate processes. These processes include aerosol formation pathways that influence the concentration of cloud condensation nuclei (CCN), with implications for Earth's albedo and climate (Charlson et al., 1987; Korhonen et al., 2008a), and the atmospheric oxidation pathways of other key climate gases, including isoprene, ammonia and organohalogens (Chen and Jang, 2012; von Glasow and Crutzen, 2004; Johnson and Bell, 2008). Thus, our ability to predict the climate into the future requires an understanding of how marine DMS production may respond to global change (Carpenter et al., 2012; Woodhouse et al., 2013; Menzo et al., 2018).
The biologically rich ice-edge regions and open seas of the Arctic are a strong source of DMS to the Arctic atmosphere (Levasseur, 2013). A seasonal cycle in CCN numbers can be related to seasonality in the Arctic DMS flux (Chang et al., 2011). Indeed, observations confirm that DMS oxidation products promote the growth of particles to produce aerosols that may influence cloud processes and atmospheric albedo (Bigg and Leck, 2001; Rempillo et al., 2011; Korhonen et al., 2008b; Chang et al., 2011). Arctic new particle formation events and peaks in aerosol optical depth (AOD) occur during summertime clean air periods (when levels of anthropogenic black carbon diminish) and have been linked to chlorophyll a maxima in surface waters and the presence of aerosols formed from DMS oxidation products such as methanesulfonate (MSA). The atmospheric oxidation products of DMS – SO2 and H2SO4 – contribute to both the growth of existing particles and new particle formation (NPF) in the Arctic atmosphere (Leaitch et al., 2013; Gabric et al., 2014; Sharma et al., 2012). Thus, the ongoing and projected rapid loss of seasonal Arctic sea ice may influence the Arctic radiation budget via changes to both the DMS flux and the associated formation and growth of cloud-influencing particles (Sharma et al., 2012). The influence that OA will have on the production and flux of DMS, and how this may further influence the Arctic radiative balance, is poorly understood and requires further experimental and modelling efforts.
During its short but highly productive summer season, the Southern Ocean is a hotspot of DMS flux to the atmosphere, influenced by the prevalence of intense blooms of DMSP-rich Phaeocystis antarctica (Schoemann et al., 2005) and the presence of persistent high winds, particularly in regions north of the sub-Antarctic front (Jarníková and Tortell, 2016). Around 3.4 Tg of sulfur is released from the Southern Ocean into the atmosphere between December and February, a flux that represents ∼15 % of global annual emissions of DMS (Jarníková and Tortell, 2016). Elevated CCN numbers are seen in the most biologically active regions of the Southern Ocean, with a significant contribution from DMS-driven secondary aerosol formation processes (McCoy et al., 2015; Korhonen et al., 2008a). DMS-derived aerosols from this region are estimated to contribute 6 to 10 W m−2 to reflected short wavelength radiation, similar to the influence of anthropogenic aerosols in the polluted Northern Hemisphere (McCoy et al., 2015). Given this important influence of polar DMS emissions on atmospheric processes and climate, it is vital we increase our understanding of the influence of future ocean acidification on DMS production.
The polar oceans are characterized by high dissolved inorganic carbon (CT) concentrations and a low carbonate system buffering capacity, mainly due to the increased solubility of CO2 in cold waters (Sabine et al., 2004; Orr et al., 2005). This makes these regions particularly susceptible to the impacts of ocean acidification (OA). For example, extensive carbonate mineral undersaturation is expected to occur in Arctic waters within the next 20–80 years (McNeil and Matear, 2008; Steinacher et al., 2009). OA has already led to a 0.1 unit decrease in global surface ocean pH, with a further fall of ∼0.4 units expected by the end of the century (Orr et al., 2005). The greatest declines in pH are likely in the Arctic Ocean, with a predicted fall of 0.45 units by 2100 (Steinacher et al., 2009), with a fall of ∼0.3 units predicted for the Southern Ocean (McNeil and Matear, 2008; Hauri et al., 2016). OA is occurring at a rate not seen on Earth for 300 Ma, and so the potential effects on marine organisms, communities and ecosystems could be wide-ranging and severe (Raven et al., 2005; Hönisch et al., 2012). Despite the imminent threat to polar ecosystems and the importance of DMS emissions to atmospheric processes, our knowledge of the response of polar DMS production to OA is limited to a single mesocosm experiment performed in a coastal fjord in Svalbard (Riebesell et al., 2013b; Archer et al., 2013) and one shipboard microcosm experiment with seawater collected from Baffin Bay (Hussherr et al., 2017). Both studies reported significant reductions in DMS concentrations with increasing levels of pCO2 during seasonal phytoplankton blooms. Hussherr et al. (2017) also saw reductions in total DMSP, whilst Archer et al. (2013) observed a significant increase in this compound, driven by CO2-induced increases in growth and abundance of dinoflagellates. However, these two single studies provide limited information on the wider response of the open Arctic or Southern oceans.
Mesocosm experiments have been a critical tool for assessing OA effects on surface ocean communities (Engel et al., 2005, 2008; Schulz et al., 2008, 2013; Hopkins et al., 2010; Webb et al., 2015, 2016; Kim et al., 2006, 2010; Crawfurd et al., 2017). The response of DMS to OA has been examined several times, predominantly at the same site in Norwegian coastal waters (Vogt et al., 2008; Hopkins et al., 2010; Webb et al., 2015; Avgoustidi et al., 2012), twice in Korean coastal waters (Kim et al., 2010; Park et al., 2014), and in a single study in the coastal Arctic waters of Svalbard (Archer et al., 2013). Mesocosm enclosures, ranging in volume from ∼11 000 to 50 000 L, allow the response of surface ocean communities to a range of CO2 treatments to be monitored under near-natural light and temperature conditions over timescales (weeks–months). This is sufficient time to allow a “winners vs. loser” dynamic to develop, whereby the succession of the phytoplankton community is altered due to the differing sensitivities of different taxonomic groups to changes in carbonate chemistry (Bach et al., 2017). The response of DMS cycling to elevated CO2 is generally driven by changes to the microbial community structure (Brussaard et al., 2013; Archer et al., 2013; Hopkins et al., 2010; Engel et al., 2008). The pseudo-natural conditions of mesocosm experiments offer the benefit of the inclusion of community dynamics of three or more trophic levels, providing the opportunity to investigate the influence of ecosystem dynamics on biogeochemical processes under experimental conditions (Riebesell et al., 2013a). Furthermore, physical processes such as particle export (Bach et al., 2016), which would be excluded by smaller-scale experiments, can be considered within the holistic mesocosm framework and make the results relevant for use within Earth system models (Six et al., 2013). However, the size, construction and associated costs of mesocosms have limited their deployment to coastal/sheltered waters, resulting in minimal geographical coverage and leaving large gaps in our understanding of the response of open ocean phytoplankton communities to OA.
Here, we adopt an alternative but complementary approach to explore the effects of OA on the cycling of DMS with the use of short-term shipboard microcosm experiments. We build on the previous temperate north-western European shelf studies of Hopkins and Archer (2014) by presenting data from four previously unpublished experiments from the north-western European shelf cruise and by extending our experimental approach to the Arctic and Southern oceans. Vessel-based research enables multiple short-term (days) near-identical incubations to be performed over extensive spatial scales that encompass natural gradients in carbonate chemistry, temperature and nutrients (Richier et al., 2014, 2018). This allows an assessment to be made of how a range of surface ocean communities, adapted to a variety of environmental conditions, respond to the same driver. The focus is then on the effect of short-term CO2 exposure on physiological processes as well as the extent of the variability in acclimation between communities. The capacity of organisms to acclimate to changing environmental conditions contributes to the resilience of key ecosystem functions, such as DMS production. Therefore, do spatially diverse communities respond differently to short-term OA, and can this be explained by the range of environmental conditions to which each is presumably already adapted? The rapid CO2 changes implemented in this study, and during mesocosm studies, are far from representative of the predicted rate of change to seawater chemistry over the coming decades, and the potential to induce a “shock” response to the sudden alteration of carbonate chemistry should be considered, particularly when working at the smaller microcosm scale. Nevertheless, our approach can provide insight into the physiological response and level of sensitivity to future OA of a variety of surface ocean communities adapted to different in situ carbonate chemistry environments (Stillman and Paganini, 2015), alongside the implications this may have for DMS production.
Communities of the north-western European shelf consistently responded to acute OA with significant increases in net DMS production, likely a result of an increase in stress-induced algal processes (Hopkins and Archer, 2014). Do polar phytoplankton communities, which are potentially adapted to contrasting biogeochemical environments, respond in the same way? By expanding our approach to encompass both polar oceans, we can assess regional contrasts in response. To this end, we combine our findings for temperate waters with those for the polar oceans into a meta-analysis to advance our understanding of the regional variability and drivers in the DMS response to OA.
2.1 Sampling stations
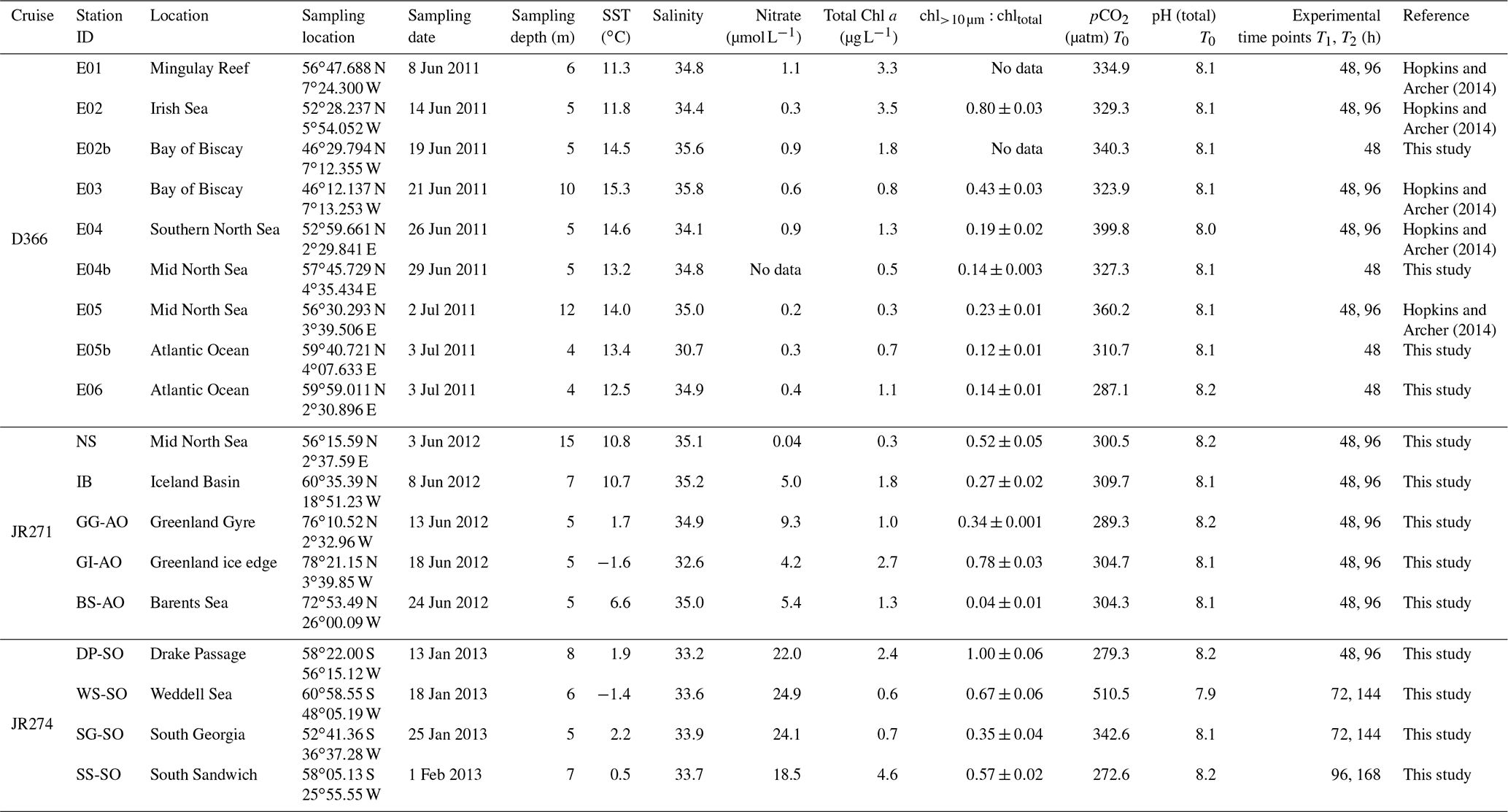
This study presents new data from two sets of field experiments carried out as a part of the UK Ocean Acidification Research Programme (UKOA) aboard the RRS James Clark Ross in the sub-Arctic and Arctic in June–July 2012 (JR271) and in the Southern Ocean in January–February 2013 (JR274). Data are combined with the results from an earlier study onboard the RRS Discovery (D366) described in Hopkins and Archer (2014) performed in the temperate waters of the north-western European shelf. Additionally, four previously unpublished experiments from D366 are also included (E02b, E04b, E05b, E06) as well as two temperate experiments from JR271 (NS and IB) (see Table 1). In total, 18 incubations were performed: 11 in temperate and sub-Arctic waters of the north-western European shelf and North Atlantic, 3 in Arctic waters and 4 in the Southern Ocean. Figure 1 shows the cruise tracks, surface concentrations of DMS and total DMSP (DMSPt) at CTD sampling stations as well as the locations of sampling for shipboard microcosms (see Table 1 for further details).
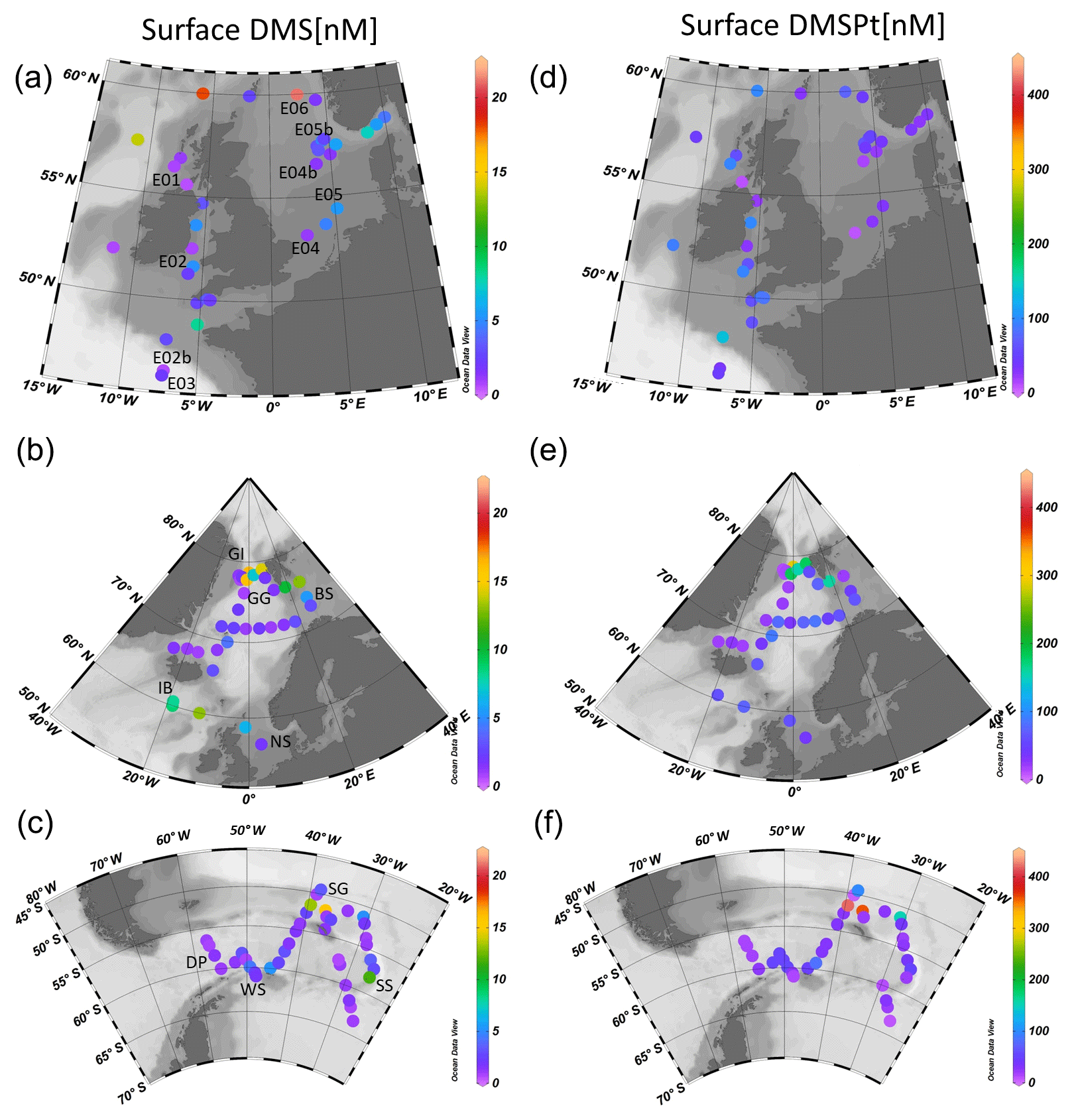
Figure 1Surface (<5 m) concentrations (nM) of DMS (a–c) and total DMSP (d–f) for cruises in the north-western European shelf (D366) (a, d), the sub-Arctic and Arctic Ocean (JR271) (b, e) and the Southern Ocean (JR274) (c, f). Locations of sampling stations for microcosm experiments shown in letters/numbers. E01–E05: see Hopkins and Archer (2014). NS: North Sea, IB: Iceland Basin, GI: Greenland Ice-edge, GG: Greenland Gyre, BS: Barents Sea, DP: Drake Passage, WS: Weddell Sea, SG: South Georgia, SS: South Sandwich.
Table 1Summary of the station locations and characteristics of the water sampled for the 18 microcosm experiments performed in temperate, sub-polar and polar waters. All polar stations were sampled for JR271 and JR274, with the exception of NS and IB.
2.2 Shipboard microcosm experiments
The general design and implementation of the experimental microcosms for JR271 and JR274 were essentially the same as for D366 and described in Richier et al. (2014, 2018) and Hopkins and Archer (2014), but with the additional adoption of trace-metal clean sampling and incubation techniques in the low trace-metal open ocean waters (see Richier et al., 2018). At each station, pre-dawn vertical profiles of temperature, salinity, oxygen, fluorescence, turbidity and irradiance were used to choose and characterize the depth of experimental water collection. Subsequently, water was collected within the mixed layer from three successive separate casts of a trace-metal clean titanium CTD rosette comprising 24 10 L Niskin bottles. Depth profiles of auxiliary measurements are shown in Fig. 2. Each cast was used to fill one of a triplicated set of experimental bottles (locations and sample depths, Table 1). Bottles were sampled within a class-100 filtered air environment within a trace-metal clean container to avoid contamination during the set-up. The water was directly transferred into acid-cleaned 4.5 L polycarbonate bottles using acid-cleaned silicon tubing, with no screening or filtration.
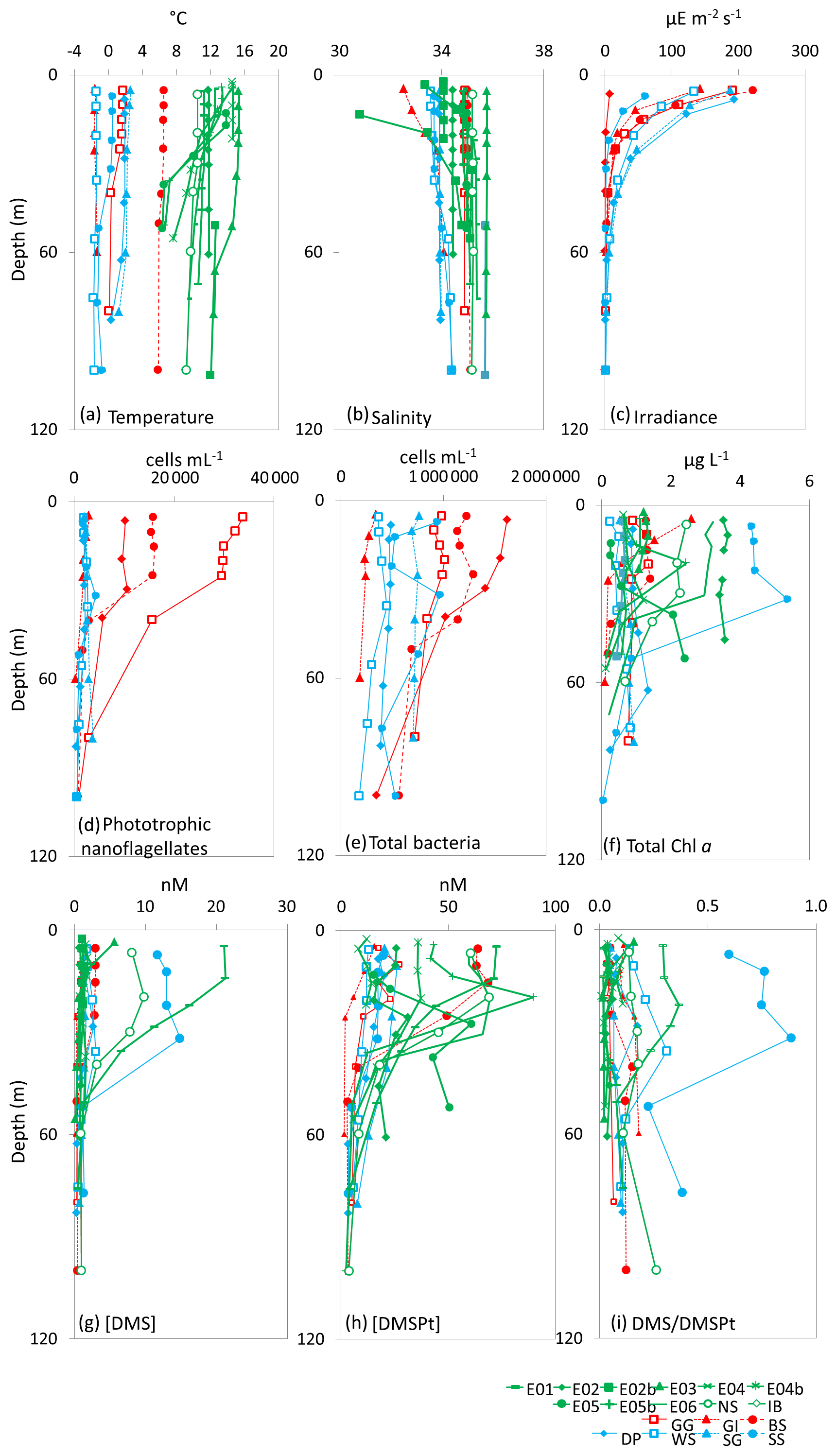
Figure 2Depth profiles down to 100 m depth for all 18 sampling stations showing (a) temperature (∘C), (b) salinity, (c) irradiance (), (d) phototrophic nanoflagellate abundance (cells mL−1), (e) total bacteria abundance (cells mL−1), (f) total Chl a (µg L−1), (g) [DMS] (nM), (h) total [DMSP] (nM) and (i) DMS/DMSPt from CTD casts at sampling stations for microcosm experiments in temperate (green), Arctic (red) and Southern Ocean (blue) waters. See Table 1 for station details. Data for irradiance, phototrophic nanoflagellates and total bacteria were not collected for temperate stations.
The carbonate chemistry within the experimental bottles was manipulated by addition of equimolar HCl and (1 mol L−1) to achieve a range of CO2 treatments: Mid CO2 (target: 550 µatm), High CO2 (target: 750 µatm), High+ CO2 (target: 1000 µatm) and High CO2 (target: 2000 µatm) (Gattuso et al., 2010). Three treatment levels were used during the sub-Arctic/Arctic microcosms (Mid, High, High+). For Southern Ocean experiments, two experiments (Drake Passage and Weddell Sea) considered one CO2 treatment (High). Three CO2 treatments (High, High+, High) were tested in the last two experiments (South Georgia and South Sandwich). Full details of the carbonate chemistry manipulations can be found in Richier et al. (2014, 2018). Broadly, achieved pCO2 levels were well-matched to target values at the start of the experiments (0 h), although differences in pCO2 between target and initial values were greater in the higher pCO2 treatments, due to lowered carbonate system buffer capacity at higher pCO2. For all 18 experiments, actual pCO2 values at 0 h were on average around 89 % ±12 % (±1 SD) of target values. The attained pCO2 values, and pCO2 at each experimental time point, are presented in Figs. 3 and 4. After first ensuring the absence of bubbles or headspace, the bottles were sealed with high-density polyethylene (HDPE) lids with silicone/polytetrafluoroethylene (PTFE) septa and placed in the incubation container. Bottles were incubated inside a custom-designed temperature- and light-controlled shipping container, set to match ( ∘C) the in situ water temperature at the time of water collection (shown in Table 1) (see Richier et al., 2018). A constant light level (100 ) was provided by daylight-simulating LED panels (Powerpax, UK). The light period within the microcosms was representative of in situ conditions. For the sub-Arctic/Arctic Ocean stations, experimental bottles were subjected to continuous light representative of the 24 h daylight of the Arctic summer. For Southern Ocean and all temperate water stations, an 18:6 light:dark cycle was used. Each bottle belonged to a set of triplicates, and sacrificial sampling of bottles was performed at two time points (see Table 1 for exact times). Use of three sets of triplicates for each time point allowed for the sample requirements of the entire scientific party (3×3 bottles, ×2 time points (see Table 1 for specific times for each experiment), ×4 CO2 treatments =72 bottles in total). Experiments were run for between 4 and 7 d (96–168 h) (15 out of 18 experiments), with initial sampling proceeded by two further time points. For three temperate experiments (E02b, E04b, E05b; see Tables 1 and 2) shorter 2 d incubations were performed, with a single sampling point at the end. E06 was run for 96 h (Tables 1 and 2). Incubation times were extended for Southern Ocean stations Weddell Sea, South Georgia and South Sandwich (see Table 1), as minimal CO2 response, attributed to slower microbial metabolism at low water temperatures, was observed for Arctic stations and the first Southern Ocean station, Drake Passage. The differential growth/metabolic rates between temperate and polar waters justify the comparison of responses of shorter-duration temperate experiments and longer-duration polar experiments. The magnitude of response was not related to incubation times, and expected differences in net growth rates (2- to 3-fold higher in temperate compared to polar waters; Eppley, 1972) did not account for the differences in response magnitude despite the increased incubation time in polar waters (see Richier et al., 2018, for detailed discussion). Samples for carbonate chemistry measurements were taken first, followed by sampling for DMS, DMSP and related parameters.
Table 2Mean (±SD) ratio of >10 µm Chl a to total Chl a () for polar microcosm sampling stations. * indicates significant difference from the response to ambient CO2. Exact CO2 treatments are down in Figs. 3 and 4.
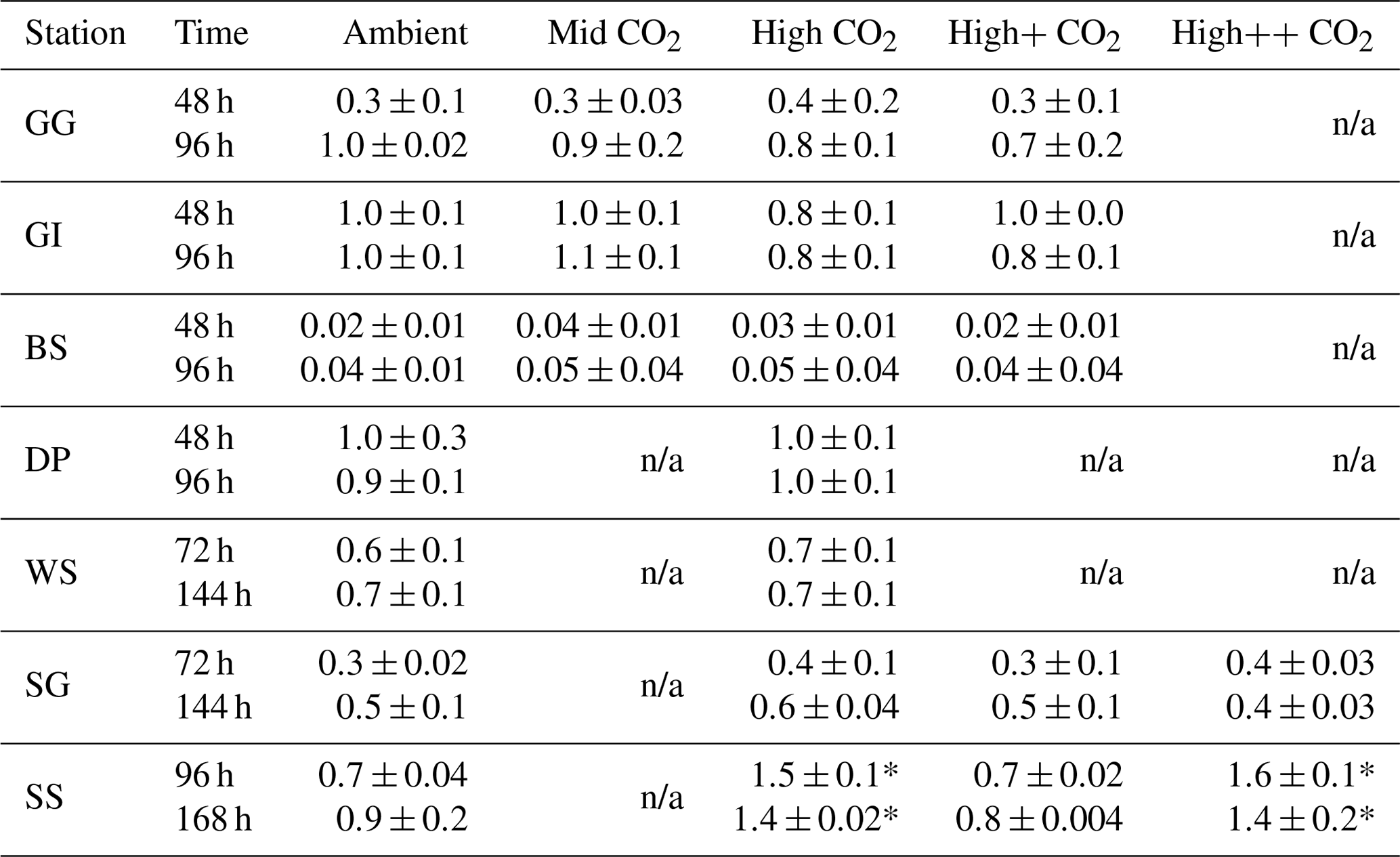
n/a: not applicable because these CO2 treatments were not included in these experiments.
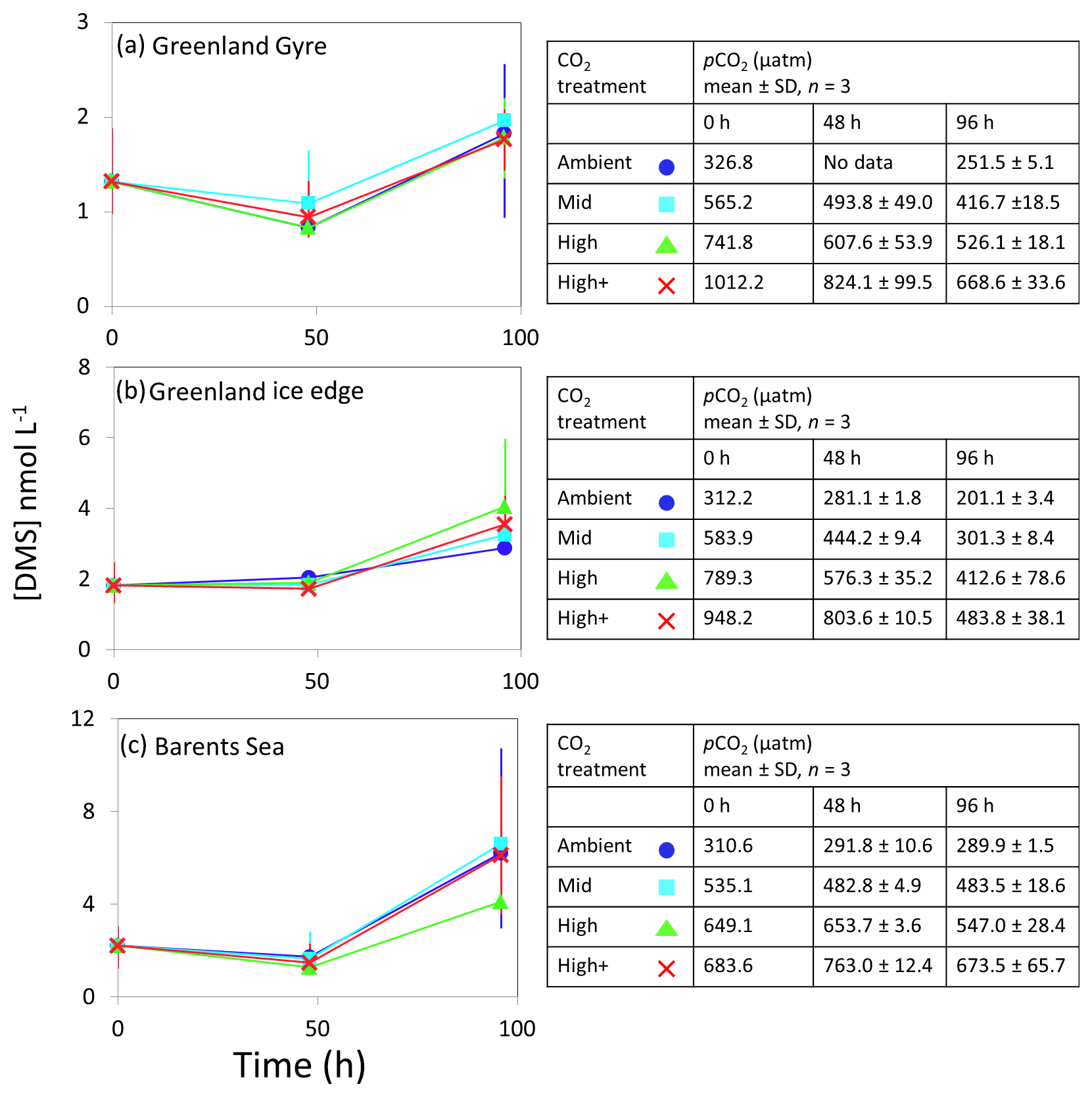
Figure 3DMS concentrations (nmol L−1) during experimental microcosms performed in Arctic waters. Data shown are the mean of triplicate incubations, and error bars show the standard error on the mean. Tables show measurements of pCO2 (µatm) for each treatment at each sampling time point. Initial measurements (0 h) were from a single sample, whilst measurements at 48 and 96 h show mean±SD of triplicate experimental bottles. Locations of water collection for microcosms shown in Fig. 1c–f.
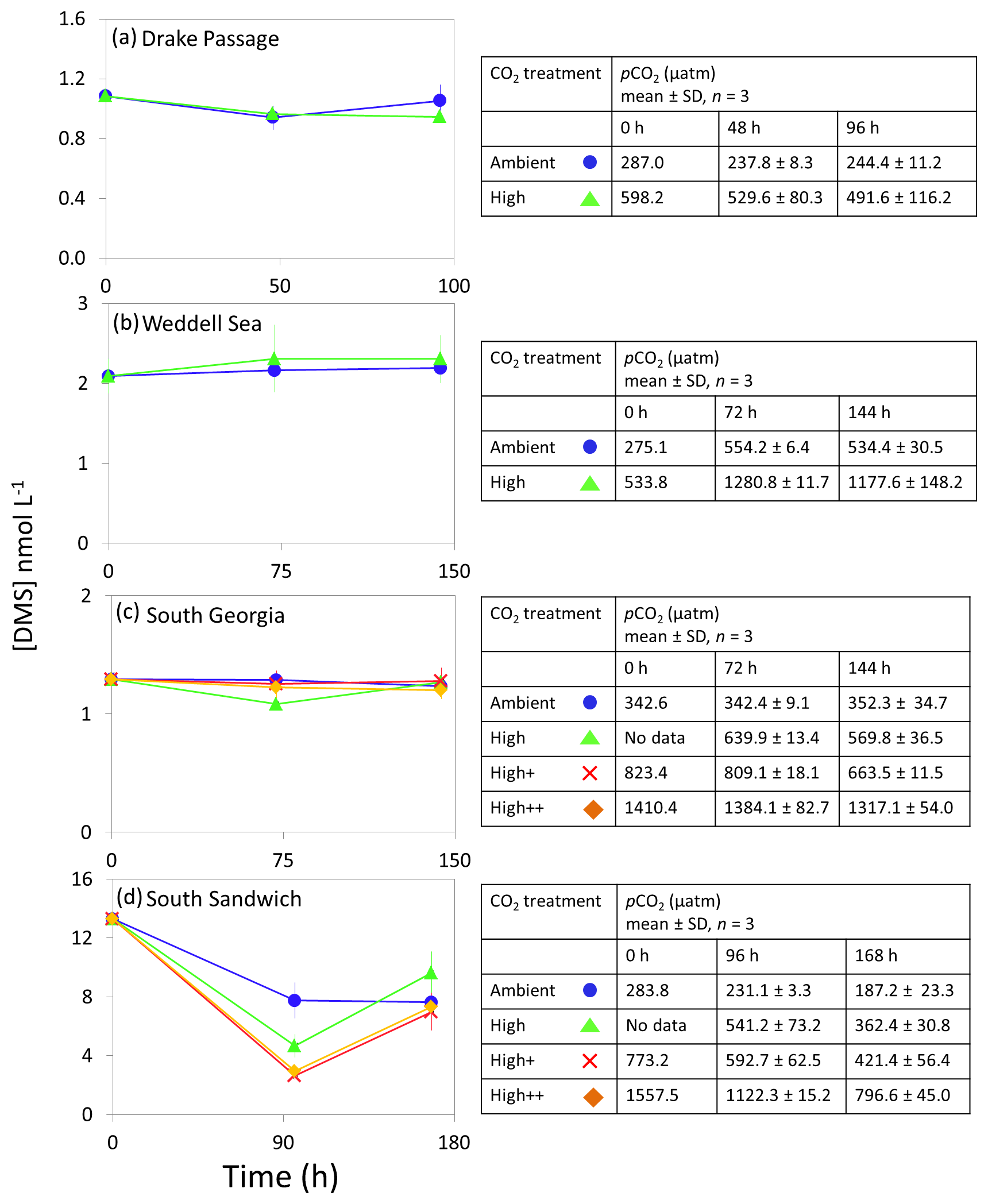
Figure 4DMS concentrations (nmol L−1) during experimental microcosms performed in Southern Ocean waters. Data shown are the mean of triplicate incubations, and error bars show the standard error on the mean. Tables show measurements of pCO2 (µatm) for each treatment at each sampling time point. Initial measurements (0 h) were from a single sample, whilst measurements at 48 and 96 h show mean±SD of triplicate experimental bottles. Locations of water collection for microcosms shown in Fig. 1c–f.
2.3 Standing stocks of DMS and DMSP
Methods for the determination of seawater concentrations of DMS and DMSP are identical to those described in Hopkins and Archer (2014) and will therefore be described in brief here. Seawater DMS concentrations were determined by cryogenic purge and trap, with gas chromatography and pulsed flame photometric detection (GC-PFPD) (Archer et al., 2013). DMSP concentrations were measured as DMS following alkaline hydrolysis. Samples for total DMSP concentrations from temperate waters were fixed by addition of 35 µL of 50 % H2SO4 to 7 mL of seawater (Kiene and Slezak, 2006) and analysed following hydrolysis within 2 months of collection (Archer et al., 2013). Samples of DMSP that were collected in polar waters were hydrolysed within 1 h of sample collection and analysed 6–12 h later. The H2SO4 fixation method was not used for samples from polar waters given the likely occurrence of Phaeocystis sp., which can result in the overestimation of DMSP concentrations (del Valle et al., 2009). Similarly, concentrations of DMSPp were determined at each time point by gravity filtering 7 mL of sample onto a 25 mm GF/F filter and preserving the filter in 7 mL of 35 mM H2SO4 in MQ water (temperate samples) or immediately hydrolysing (polar samples) and analysing by GC-PFPD. DMS calibrations were performed using alkaline cold hydrolysis (1 M NaOH) of DMSP sequentially diluted three times in MilliQ water to give working standards in the range 0.03–3.3 ng S mL−1. Five point calibrations were performed every 2–4 d throughout the cruise.
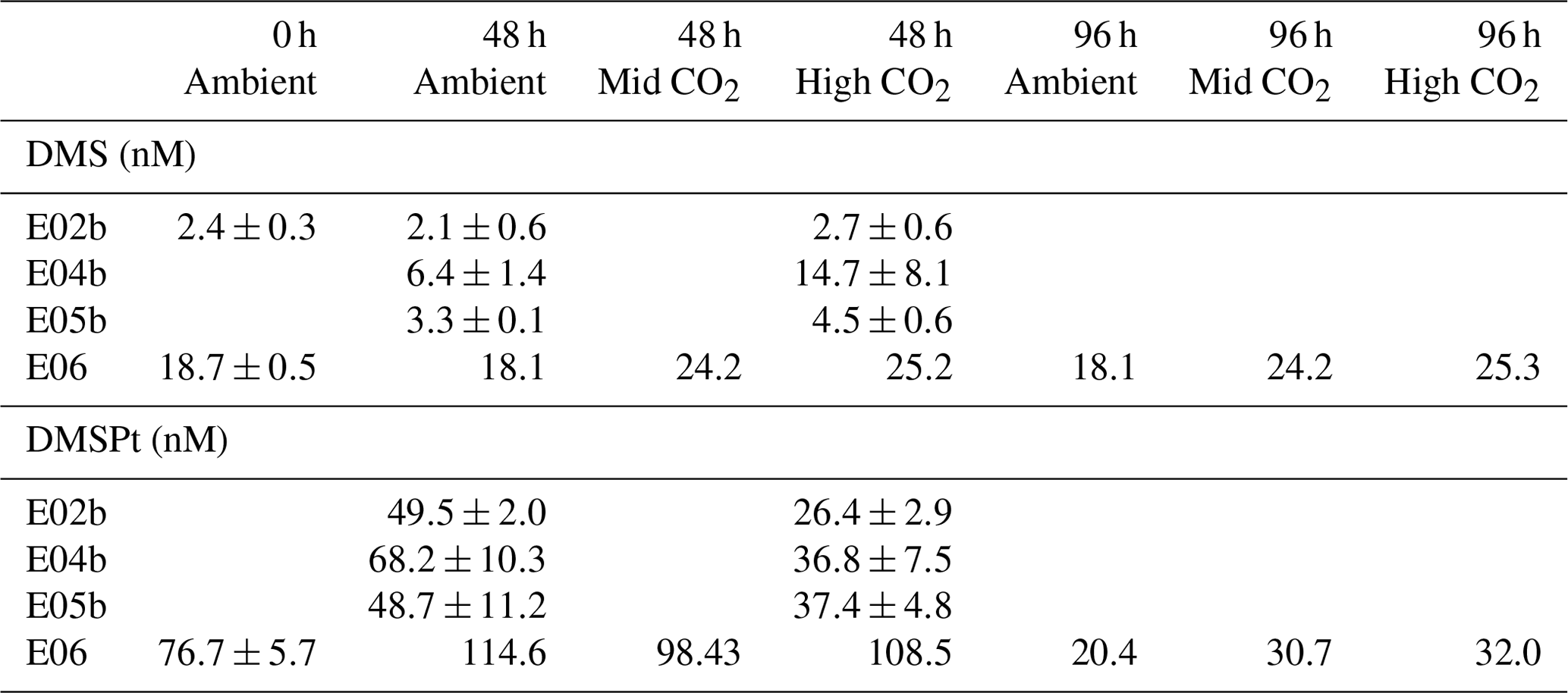
Table 3DMS and DMSPt response (mean±SD, n=3) to high CO2 treatments during previously unpublished small-scale experiments from north-western European shelf cruise D366. For details of the sampling stations, see Table 1.
2.4 De novo DMSP synthesis
De novo DMSP synthesis and gross production rates were determined for all microcosm experiments, except Barents Sea and South Sandwich, at each experimental time point, using methods based on the approach of Stefels et al. (2009) and described in detail in Archer et al. (2013) and Hopkins and Archer (2014). Triplicate rate measurements were determined for each CO2 level. For each rate measurement three ×500 mL polycarbonate bottles were filled by gently siphoning water from each replicate microcosm bottle. Trace amounts of NaH13CO3, equivalent to ∼6 % of in situ dissolved inorganic carbon (CT), were added to each 500 mL bottle. The bottles were incubated in the microcosm incubation container with temperature and light levels as described earlier. Samples were taken at 0 h, and then at two further time points over a 6–9 h period. At each time point, 250 mL was gravity filtered in the dark through a 47 mm GF/F filter, the filter gently folded and placed in a 20 mL serum vial with 10 mL of Milli-Q and one NaOH pellet, and the vial crimp-sealed. Samples were stored at −20 ∘C until analysis by a proton transfer reaction-mass spectrometer (PTR-MS) (Stefels et al., 2009).
The specific growth rate of DMSP (μDMSP) was calculated assuming exponential growth from
(Stefels et al., 2009), where 64MPt, 64MPt−1, and 64MPt+1 are the proportion of 1×13C labelled DMSP relative to total DMSP at time t, at the preceding time point (t−1) and at the subsequent time point (t+1), respectively. Values of 64MP were calculated from the protonated masses of DMS as mass , determined by PTR-MS. 64MPeq is the theoretical equilibrium proportion of 1×13C based on a binomial distribution and the proportion of tracer addition. An isotope fractionation factor αk of 1.06 is included, based on laboratory culture experiments using Emiliania huxleyi (Stefels et al., 2009). In vivo DMSP gross production rates during the incubations () were calculated from μDMSP and the initial particulate DMSP (DMSPp) concentration of the incubations (Hopkins and Archer, 2014; Stefels et al., 2009). These rates provide important information on how the physiological status of DMSP-producing cells may be affected by OA within the bioassays.
2.5 Seawater carbonate chemistry analysis
The techniques and methods used to determine both the in situ and experimental carbonate chemistry parameters, and to manipulate seawater carbonate chemistry within the microcosms, are described in Richier et al. (2014) and will only be given in brief here. Experimental T0 measurements were taken directly from CTD bottles and immediately measured for total alkalinity (AT) (Apollo SciTech AS-Alk2 Alkalinity Titrator) and dissolved inorganic carbon (CT) (Apollo SciTech CT analyser (AS-C3) with LICOR 7000). The CO2SYS program (version 1.05) (Lewis and Wallace, 1998) was used to calculate the remaining carbonate chemistry parameters including pCO2.
Measurements of AT and CT were made from each bottle at each experimental time point and again used to calculate the corresponding values for pCO2 and pHT. The carbonate chemistry data for each sampling time point for each experiment are summarized in Tables S1, S2 and S3 in the Supplement (experimental starting conditions are given in Table 1).
2.6 Chlorophyll a (Chl a) determinations
Concentrations of Chl a were determined as described in Richier et al. (2014). Briefly, 100 mL aliquots of seawater from the incubation bottles were filtered through either 25 mm GF/F (Whatman, 0.7 µm pore size) or polycarbonate filters (Whatman, 10 µm pore size) to yield total and >10 µm size fractions, with the <10 µm fraction calculated by the difference. Filters were extracted in 6 mL HPLC-grade acetone (90 %) overnight in a dark refrigerator. Fluorescence was measured using a Turner Designs Trilogy fluorometer, which was regularly calibrated with dilutions of pure Chl a (Sigma, UK) in acetone (90 %).
2.7 Community composition
Small phytoplankton community composition was assessed by flow cytometry. For details of the methodology, see Richier et al. (2014).
2.8 Data handling and statistical analyses
Permutational analysis of variance (PERMANOVA) was used to analyse the difference in response of DMS and DMSP concentrations to OA, both between and within the two polar cruises in this study. Both dependent variables were analysed separately using a nested factorial design with three factors: (i) cruise locations: Arctic and Southern oceans, (ii) experiment location nested within the cruise location (see Table 1 for station IDs) and (iii) CO2 level: 385, 550, 750, 1000 and 2000 µatm. The main effects and pairwise comparisons of the different factors were analysed through unrestricted permutations of raw data. If a low number of permutations were generated, then the p value was obtained through random sampling of the asymptotic permutation distribution using Monte Carlo tests.
One-way analysis of variance was used to identify differences in the ratio of >10 µm Chl a to total Chl a (chl>10 µm: chltot; see Discussion). Initially, tests of normality were applied ( not normal), and if data failed to fit the assumptions of the test, linearity transformations of the data were performed (logarithmic or square root) and the ANOVA proceeded from this point. The results of ANOVA are given as follows: F: ratio of mean squares, df: degrees of freedom, p: level of confidence. For those data still failing to display normality following transformation, a rank-based Kruskal–Wallis test was applied (H: test statistic, df: degrees of freedom, p: level of confidence).
3.1 Sampling stations
At temperate sampling stations, sea surface temperatures ranged from 10.7 ∘C for Iceland Basin to 15.3 ∘C for Bay of Biscay, with surface salinity in the range 34.1–35.2, with the exception of station E05b, which had a relatively low salinity of 30.5 (Fig. 2 and Table 1). Seawater temperatures at the polar microcosm sampling stations ranged from −1.5 ∘C at sea-ice influenced stations (Greenland Ice-edge and Weddell Sea) up to 6.5 ∘C for Barents Sea (Fig. 2a). Salinity values at all the Southern Ocean stations were <34, whilst they were ∼35 at all the Arctic stations, with the exception of Greenland Ice-edge, which had the lowest salinity of 32.5 (Fig. 2b). Phototrophic nanoflagellate abundances were variable, with cells mL−1 at Greenland Gyre, 1.5×104 cells mL−1 at Barents Sea and cells mL−1 for all other stations (Fig. 2d). Total bacterial abundances ranged from 3×105 cells mL−1 at Greenland Ice-edge up to 3×106 cells mL−1 at Barents Sea (Fig. 2e).
Chl a concentrations in temperate waters ranged from 0.3 µg L−1 for two North Sea stations (E05 and North Sea) up to 3.5 µg L−1 for Irish Sea (Fig. 2 and Table 1). Chl a was also variable in polar waters, exceeding 4 µg L−1 at South Sandwich and 2 µg L−1 at Greenland Ice-edge, whilst the remaining stations ranged from 0.2 µg L−1 (Weddell Sea) to 1.5 µg L−1 (Barents Sea) (Fig. 2). The high Chl a concentrations at South Sandwich correspond to low in-water irradiance levels at this station (Fig. 2c).
In temperate waters, maximum DMS concentrations were generally seen in near-surface measurements, ranging from 1.0 nmol L−1 for E04 to 21.1 nmol L−1 for E06, with rapidly decreasing concentrations with depth (Fig. 2g). As an exception to this, DMS concentrations at South Sandwich showed a sub-surface maximum of 15 nM at 32 m, coincident with a subsurface Chl a maximum of 5.4 µg L−1. DMSP generally ranged from 12 to 20 nmol L−1, except Barents Sea, where surface concentrations exceeded 60 nmol L−1 (Fig. 2h). DMSP tended to peak in the near-surface waters, ranging from 12.0 nmol L−1 for E04 to 72.5 nmol L−1 for E06, although in some cases a subsurface maximum in overall DMSP concentrations was seen, as observed for E05b (89.8 nmol L−1 20 m), and again coincident with a subsurface Chl a peak of >2 µg L−1 (Fig. 2f and h). Surface DMS concentrations in polar waters were generally lower than temperate waters, ranging from 1 to 3 nmol L−1, with the exception of South Sandwich, where concentrations of ∼12 nmol L−1 were observed (Fig. 2g), and resulted in a high DMS:DMSP of 0.6–0.9 in the surface layer (Fig. 2i). DMS:DMSP did not exceed 0.5 at any other sampling stations.
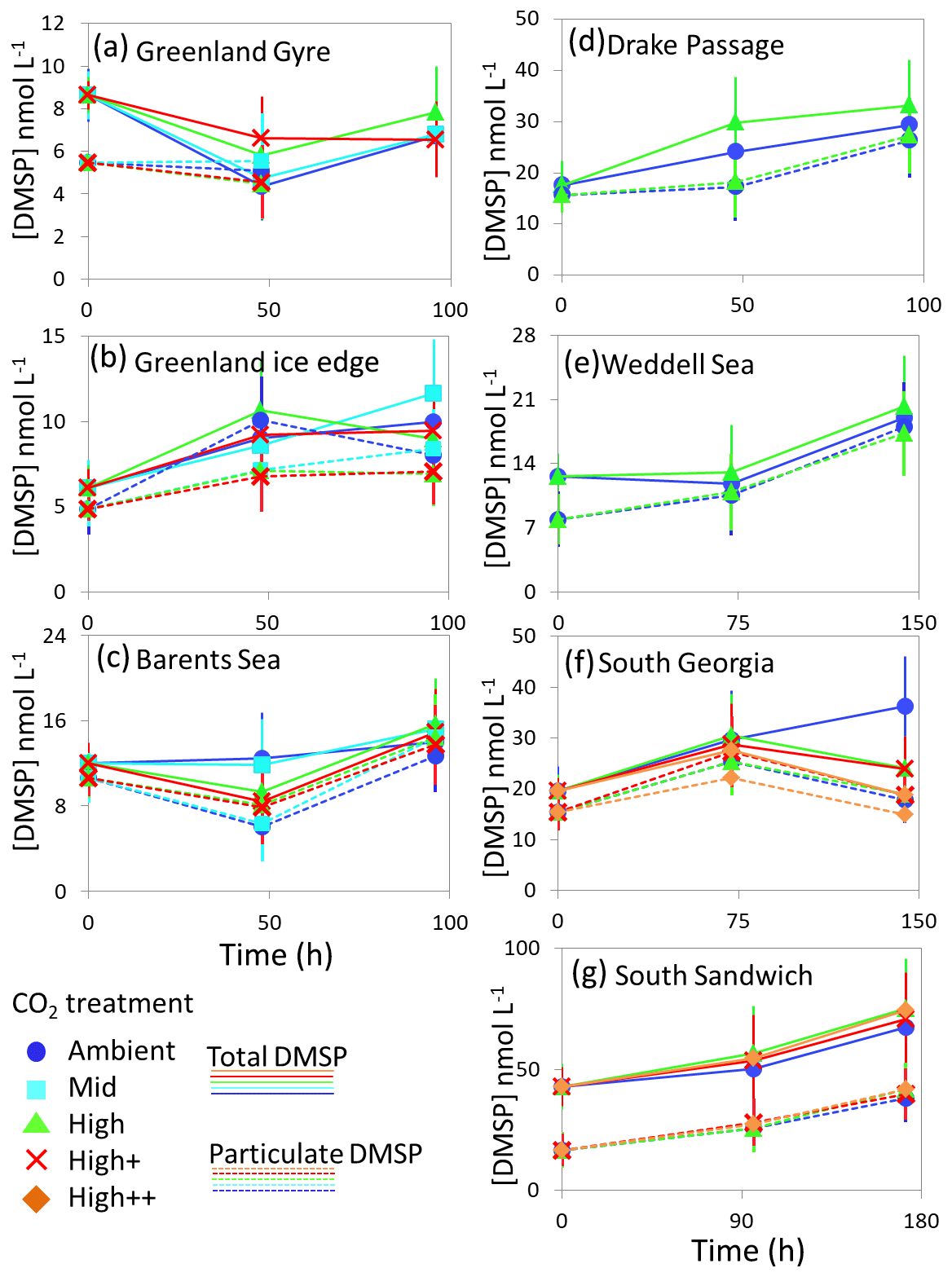
Figure 5Total DMSP (solid lines) and particulate DMSP (dashed lines) concentrations (nmol L−1) during experimental microcosms performed in Arctic waters (a–c) and in Southern Ocean waters (d–g). Data shown are the mean of triplicate incubations, and error bars show the standard error on the mean. Locations of water collection for microcosms shown in Fig. 1c–f. Particulate DMSP concentrations were used in calculations of DMSP production rates (Fig. 6).
3.2 Response of DMS and DMSP to OA
The temporal trend in DMS concentrations showed a similar pattern for the three Arctic Ocean experiments. Initial concentrations of 1–2 nmol L−1 remained relatively constant over the first 48 h and then showed small increases of 1–4 nmol L−1 over the remainder of the incubation period (Fig. 3). Increased variability between triplicate incubations became apparent in all three Arctic experiments by 96 h, but no significant effects of elevated CO2 on DMS concentrations were observed. Initial DMSP concentrations were more variable, from 6 nmol L−1 at Greenland Ice-edge to 12 nmol L−1 at Barents Sea, and either decreased slightly (net loss 1–2 nmol L−1 GG) or increased slightly (net increase ∼4 nmol L−1 Greenland Ice-edge, ∼3 nmol L−1 Barents Sea) (Fig. 5a–c). DMSP concentrations were found to decrease significantly in response to elevated CO2 after 48 h for Barents Sea (Fig. 5c, t=2.05, p=0.025), whilst no significant differences were seen after 96 h. No other significant responses in DMSP were identified.
The range of initial DMS concentrations was greater at Southern Ocean sampling stations compared to the Arctic, from 1 nmol L−1 at Drake Passage up to 13 nmol L−1 at South Sandwich (Fig. 4). DMS concentrations showed little change over the course of 96–168 h incubations and no effect of elevated CO2, with the exception of South Sandwich (Fig. 4d). Here, concentrations decreased sharply after 96 h by between 3 and 11 nmol L−1. Concentrations at 96 h were CO2-treatment dependent, with significant decreases in DMS concentration occurring with increasing levels of CO2 (PERMANOVA, t=2.61, p=0.028). Significant differences ceased to be detectable by the end of the incubations (168 h). Initial DMSP concentrations were higher at the Southern Ocean stations than for Arctic stations, ranging from 13 nmol L−1 for Weddell Sea to 40 nmol L−1 for South Sandwich (Fig. 5d–g). Net increases in DMSP occurred throughout, except at South Georgia, and were of the order of between <10 and >30 nmol L−1 over the course of the incubations. Concentrations were not generally pCO2-treatment dependent, with the exception of the final time point at South Georgia (144 h), when a significantly lower DMSP with increasing CO2 was observed (PERMANOVA, , p<0.001).
Results from the previously unpublished experiments from temperate waters are in strong agreement with the five experiments presented in Hopkins and Archer (2014), with consistently decreased DMS concentrations and enhanced DMSP under elevated CO2. The data are presented in Table 3 and Fig. S2 in the Supplement and included in the meta-analysis in Sect. 4.1 of this paper.
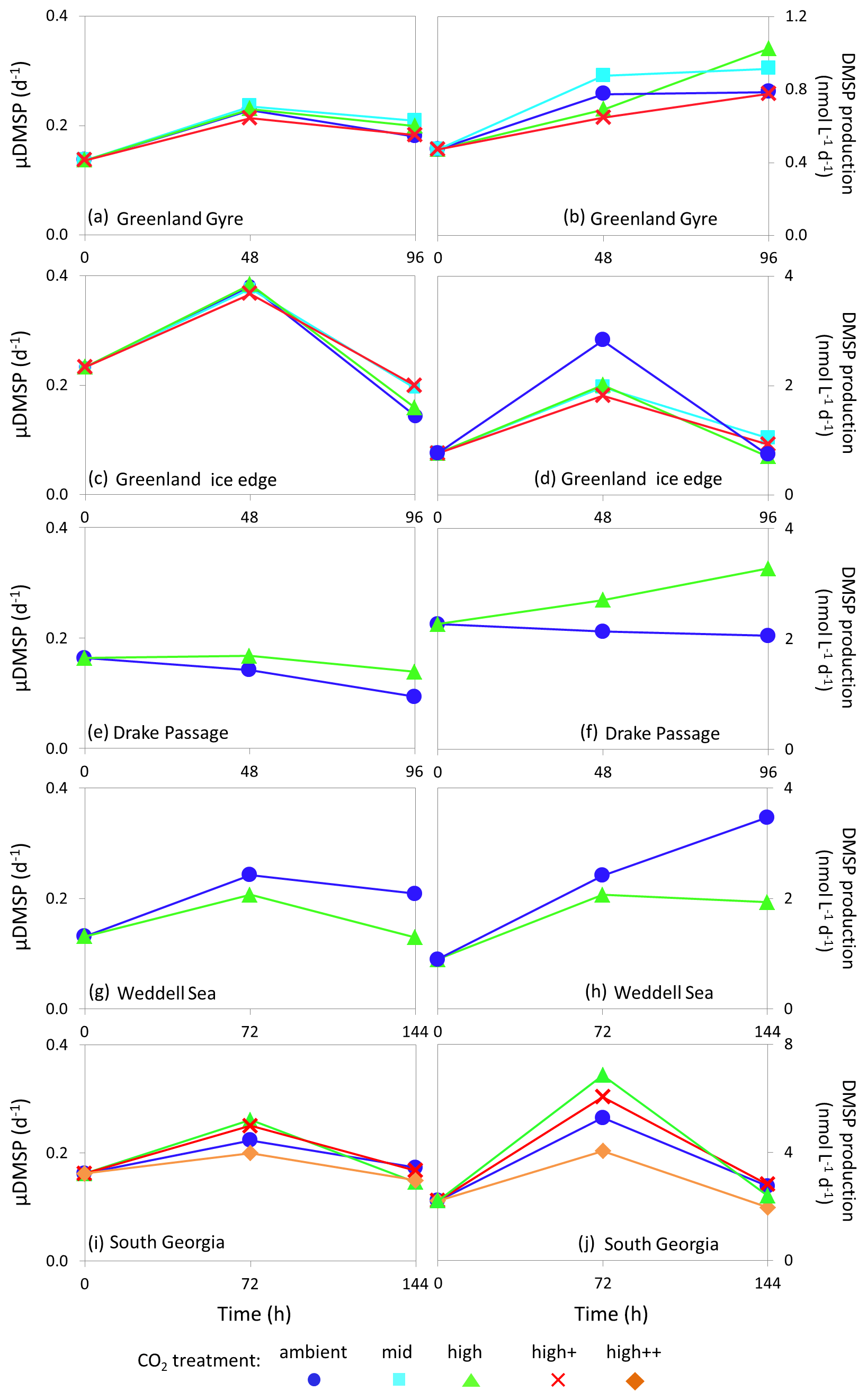
Figure 6De novo synthesis of DMSP (μDMSP, d−1) (left column) and DMSP production rates () (right column) for Arctic Ocean stations Greenland Gyre (a, b) and Greenland Ice-edge (c, d) and Southern Ocean stations Drake Passage (e, f), Weddell Sea (g, h) and South Georgia (i, j). No data are available for Barents Sea (Arctic Ocean) or South Sandwich (Southern Ocean).
3.3 Response of de novo DMSP synthesis and production to OA
Rates of de novo DMSP synthesis (μDMSP) at initial time points ranged from 0.13 d−1 (Weddell Sea, Fig. 6g) to 0.23 d−1 (Greenland Ice-edge, Fig. 6c), whilst DMSP production ranged from 0.4 (Greenland Gyre, Fig. 6b) to 2.27 (Drake Passage, Fig. 6f). Maximum rates of μDMSP of 0.37–0.38 d−1 were observed at Greenland Ice-edge after 48 h of incubation in all CO2 treatments (Fig. 6c). The highest rates of DMSP production were observed at South Georgia after 96 h of incubation and ranged from 4.1 to 6.9 across CO2 treatments (Fig. 6j). Rates of DMSP synthesis and production were generally lower than those measured in temperate waters (Hopkins and Archer, 2014) (initial rates: μDMSP 0.33–0.96 d−1, 7.1–37.3 ), but were comparable to measurements made during an Arctic mesocosm experiment (Archer et al., 2013) (0.1–0.25 d−1, 3–5 in non-bloom conditions). The lower rates in cold polar waters likely reflect slower metabolic processes and are reflected by standing stock DMSP concentrations which were also lower than in temperate waters (5–40 nmol L−1 polar, 8–60 nmol L−1 temperate; Hopkins and Archer, 2014). No consistent effects of high CO2 were observed for either DMSP synthesis or production in polar waters, similar to findings for DMSP standing stocks. However, some notable but contrasting differences between CO2 treatments were observed. There were 36 % and 37 % increases in μDMSP and DMSP production, respectively, at 750 µatm for Drake Passage after 96 h (Fig. 6e, f) and 38 % and 44 % decreases in both at 750 µatm after 144 h for Weddell Sea (Fig. 5g, h). For Drake Passage, the difference between treatments at 96 h coincided with significantly higher nitrate concentrations in the high CO2 treatment (nitrate/nitrite at 96 h: ambient = 18.9±0.2 µmol L−1, µmol L−1, ANOVA F=62.619, df=1, p=0.001). However, it is uncertain whether the difference in nutrient availability between treatments (approximately 5 %) would be significant enough to strongly influence the rate of DMSP production.
The differences in DMSP production rates did not correspond to any other measured parameter. It is possible that changes in phytoplankton community composition may have led to differences in DMSP production rates for Drake Passage and Weddell Sea, but no quantification of large cells (diatoms, dinoflagellates) was undertaken for these experiments.
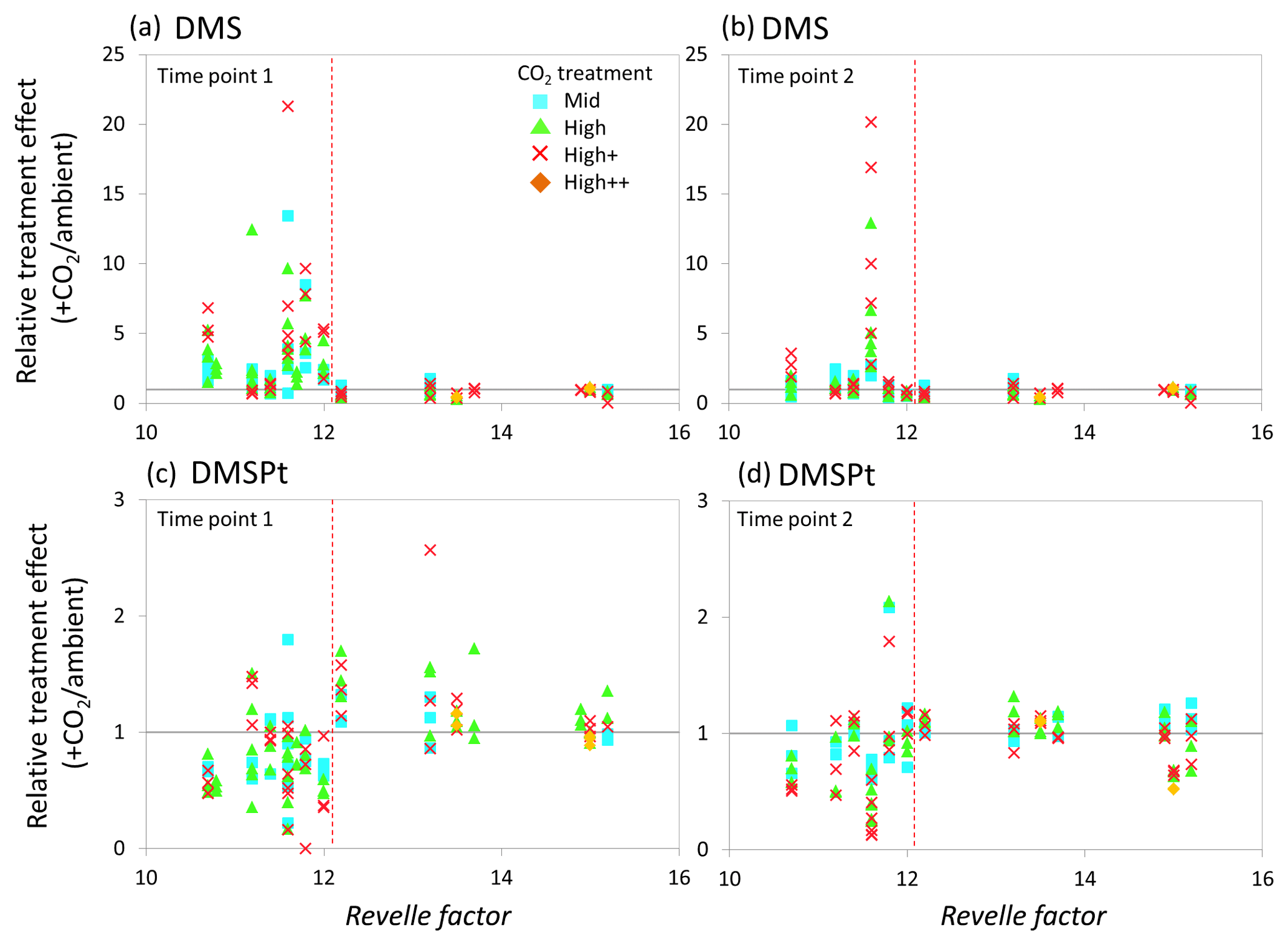
Figure 7Relationship between the Revelle factor of the sampled water and the relative CO2 treatment effect at for concentrations of DMS at time point 1 (a) and time point 2 (b) and for total DMSP concentrations at time point 1 (c) and time point 2 (d) for all microcosm experiments performed in north-western European waters, sub-Arctic and Arctic waters, and the Southern Ocean. Grey solid line (=1) indicates no effect of elevated CO2. Revelle factor >12: polar waters (indicated by red dashed line). Time point 1=48 h, except for WS and SG (72 h) and SS (96 h). For detailed analyses of the north-western European shelf data, see Hopkins and Archer (2014).
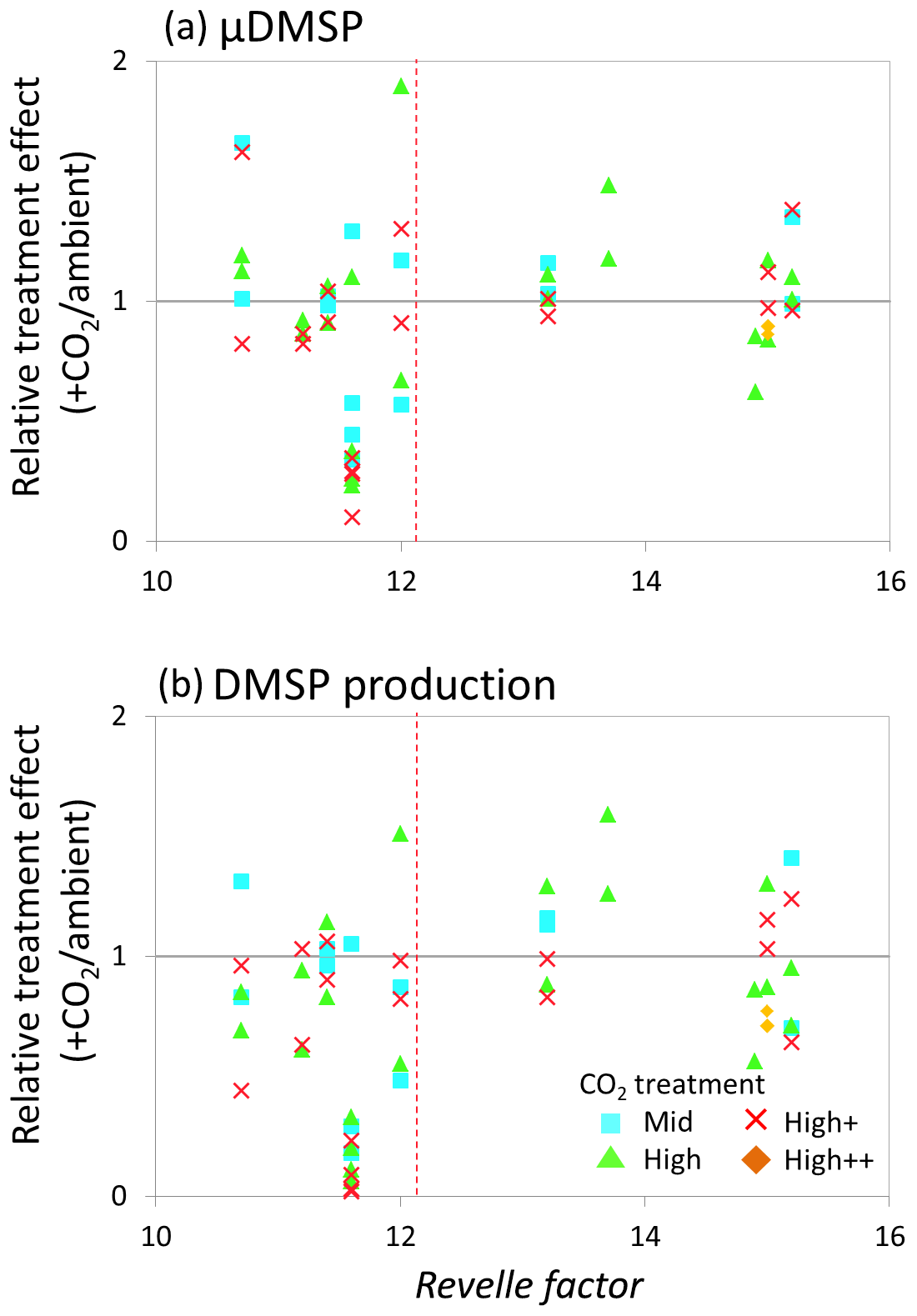
Figure 8Relationship between the Revelle factor of the sampled water and the relative CO2 treatment effect at for (a) de novo DMSP synthesis (μDMSp, d−1) at both time points 1 and 2 and (b) DMSP production rate () at both time points 1 and 2 for microcosm experiments performed in north-western European waters, sub-Arctic and Arctic waters, and the Southern Ocean. Grey solid line (=1) indicates no effect of elevated CO2. Revelle factor >12: polar waters (indicated by red dashed line). Time point 1=48 h and time point 2=96 h, except for Weddell Sea and South Georgia (1=72 h, 2=144 h). For discussion of the north-western European shelf data, see Hopkins and Archer (2014).
4.1 Regional differences in the response of DMS(P) to OA
We combine our findings from the polar oceans with those from temperate waters into a meta-analysis in order to assess the regional variability and drivers in the DMS(P) response to OA. Figures 7 and 8 provide an overview of the results discussed so far in this current study, together with the results from Hopkins and Archer (2014) as well as the results from four previously unpublished microcosm experiments from the north-western European shelf cruise and a further two temperate water microcosm experiments from the Arctic cruise (North Sea and Iceland Basin, Table 1). This gives a total of 18 microcosm experiments, each with between one and three high CO2 treatments.
Hopkins and Archer (2014) reported consistent and significant increases in DMS concentration in response to elevated CO2 that were accompanied by significant decreases in DMSPt concentrations. Bacterially mediated DMS processes appeared to be insensitive to OA, with no detectable effects on dark rates of DMS consumption and gross production and no consistent response seen in bacterial abundance (Hopkins and Archer, 2014). In general, there were large short-term decreases in Chl a concentrations and phototrophic nanoflagellate abundance in response to elevated CO2 in these experiments (Richier et al., 2014).
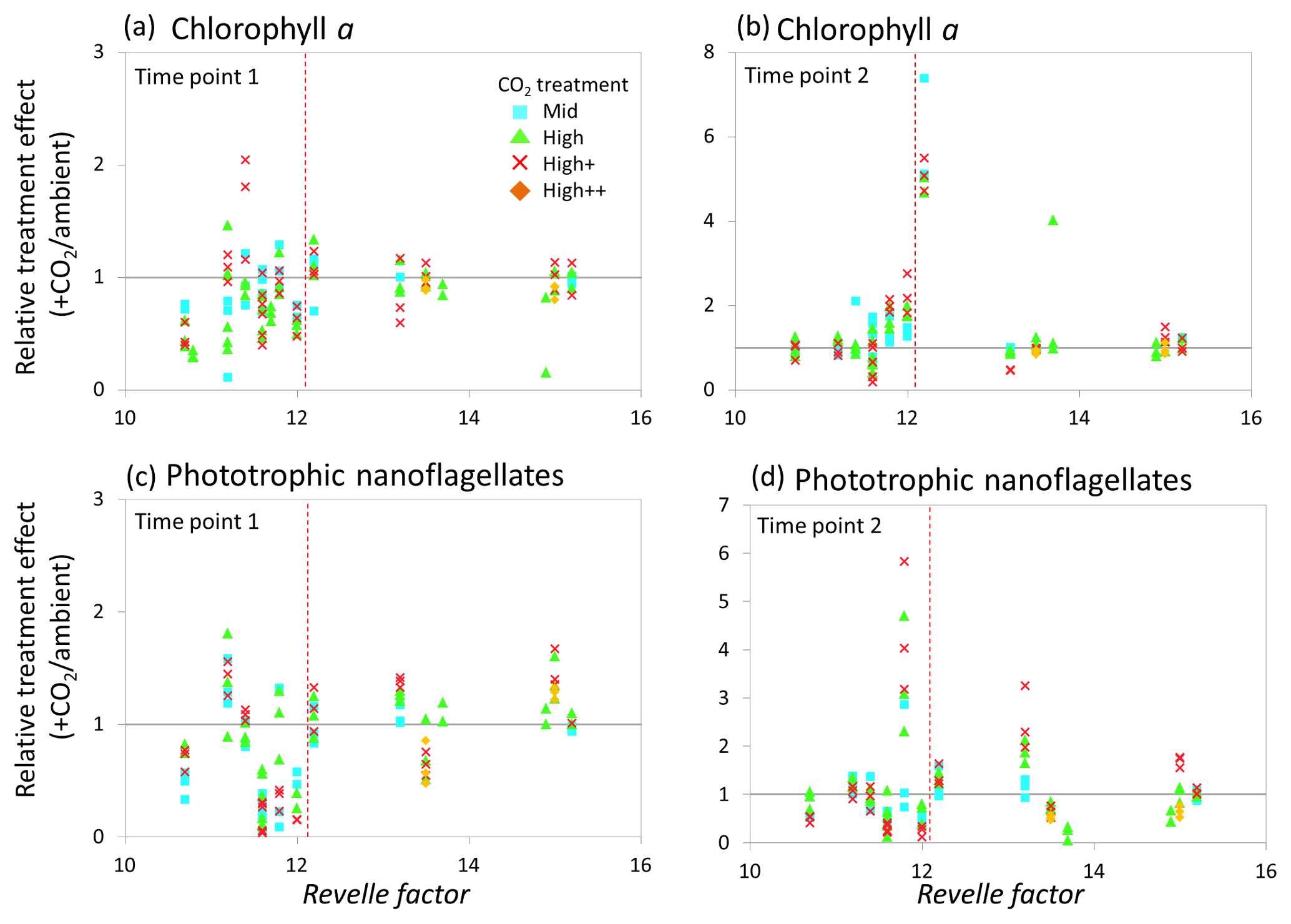
Figure 9Relationship between the Revelle factor of the sampled water and the relative CO2 treatment effect for chlorophyll a concentrations at time point 1 (a) and time point 2 (b) and phototrophic nanoflagellate abundance at time point 1 (c) and time point 2 (d) for all microcosm experiments performed in north-western European waters, sub-Arctic and Arctic waters, and the Southern Ocean. Grey solid line (=1) indicates no effect of elevated CO2. Revelle factor >12: polar waters (indicated by red dashed line). T1=48 h and T2=96 h, except for Weddell Sea and South Georgia (72, 144 h) and South Sandwich (96, 168 h).
The relative treatment effects () for DMS and DMSP (Fig. 7), DMSP synthesis and production (Fig. 8), and Chl a and phototrophic nanoflagellate abundance (Fig. 9) are plotted against the Revelle factor of the sampled waters. The Revelle factor (R), calculated here with CO2Sys using measurements of carbonate chemistry parameters (, Lewis and Wallace, 1998), describes how the partial pressure of CO2 in seawater (PCO2) changes for a given change in DIC (Sabine et al., 2004; Revelle and Suess, 1957). Its magnitude varies latitudinally, with lower values (9–12) from the tropics to temperate waters and the highest values in cold high-latitude waters (13–15). Thus polar waters can be considered poorly buffered with respect to changes in DIC. Therefore, biologically driven seasonal changes in seawater pCO2 would result in larger changes in pH than would be experienced in temperate waters (Egleston et al., 2010). Furthermore, the seasonal sea-ice cycle strongly influences carbonate chemistry, such that sea-ice regions exhibit wide fluctuations in carbonate chemistry (Revelle and Suess, 1957; Sabine et al., 2004). Sampling stations with a R above ∼12 represent the seven polar stations (right of the red dashed line in Figs. 7, 8, and 9). The surface waters of the polar oceans have naturally higher levels of DIC and a reduced buffering capacity, driven by higher CO2 solubility in colder waters (Sabine et al., 2004). Thus, the relationship between experimental response and R is a simple way of demonstrating the differences in response to OA between temperate and polar waters and provides some insight into how the CO2 sensitivity of different surface ocean communities may relate to the in situ carbonate chemistry. The effect of elevated CO2 on DMS concentrations at polar stations, relative to ambient controls, was minimal at both sampling points and is in strong contrast to the results from experiments performed in waters with lower values of R on the north-western European shelf. In contrast, at temperate stations, DMSP concentrations displayed a clear negative treatment effect, whilst at polar stations a positive effect was evident under high CO2 and particularly at the first time point (48–96 h) (Fig. 7c and d). De novo DMSP synthesis and DMSP production rates show a less consistent response in either environment (Fig. 8a and b), although a significant suppression of DMSP production rates in temperate waters compared to polar waters was seen (Fig. 8b, Kruskal–Wallis one-way ANOVA H=8.711, df=1, p=0.003). A similar but not significant response was seen for de novo DMSP synthesis (Fig. 8a).
Our data imply that DMSP concentrations in temperate waters were downregulated in response to OA, attributed to the adverse effects of rapid OA on the growth of DMSP producers which led to reductions in the abundance of these types of phytoplankton (Richier et al., 2014; Hopkins and Archer, 2014). By comparison, a more muted, but generally positive, DMSP response was seen in polar waters at the first time point, whilst these treatment effects were more or less undetectable by the second time point. There is some evidence that the enhanced DMSP concentrations in polar waters were accompanied by increased DMSP production rates (Fig. 8), although data are not available for all the experiments. However, these changes may reflect a short-term “shock” physiological protective response to the experimental OA, similar to that seen in response to other short-term stressors such as high irradiance that result in an increase in DMSP concentrations (Sunda et al., 2002; Galindo et al., 2016). The lack of a treatment effect in DMSP concentrations by the second time point may be indicative that the community had, to some extent, acclimated to the change, allowing DMSP production/concentrations to return to baseline levels. This may reflect a higher degree of tolerance to rapid changes in carbonate chemistry amongst polar communities – species which are already adapted to highly variable irradiance/carbonate chemistry regimes (Thomas and Dieckmann, 2002; Rysgaard et al., 2012; Thoisen et al., 2015). Further experiments with polar communities would help to unravel the potential importance of such mechanisms and whether they facilitated the ability of polar phytoplankton communities to resist the high CO2 treatments.
The responses to OA observed for DMS and DMSP production are likely to be reflected in the dynamics of the DMSP-producing phytoplankton. In an assessment across all experiments, Richier et al. (2018) showed that the magnitude of biological responses to short-term CO2 changes reflected the buffer capacity of the sampled waters. A consistent suppression of net growth rates in small phytoplankton (<10 µm) and total Chl a concentrations was observed under high CO2 within experiments performed in temperate waters with a higher buffer capacity.
Generally, less significant relationships were found between the phytoplankton response and the other wide range of physical, chemical or biological variables that were examined (Richier et al., 2018).
In correspondence to the analyses carried out by Richier et al. (2018), at 48–96 h (see Table 1), a statistically significant difference in response was seen between temperate and polar waters for Chl a (Kruskal–Wallis one-way ANOVA H=20.577, df=1, p<0.001). In general, at polar stations phytoplankton showed minimal response to elevated CO2, in contrast to a strong negative response in temperate waters (Fig. 9a). By the second time point (96–144 h; see Table 1), no significant difference in response of Chl a between temperate and polar waters was apparent (Fig. 9b). As shown in Richier et al. (2014), phototrophic nanoflagellates responded to high CO2 with large decreases in abundance in temperate waters and increases in abundance in polar waters (Fig. 9c and d), with some exceptions: North Sea and South Sandwich gave the opposite response. The responses had lessened by the second time point (96–168 h; see Table 1).
In contrast, bacterial abundance did not show the same regional differences in response to high CO2 (see Hopkins and Archer, 2014, for temperate waters, and Fig. S1 in the Supplement for polar waters). Bacterial abundance in temperate waters gave variable and inconsistent responses to high CO2. For all Arctic stations, as well as Southern Ocean stations Drake Passage and Weddell Sea, no response to high CO2 was observed. For South Georgia and South Sandwich, bacterial abundance increased at 1000 and 2000 µatm, with significant increases for South Georgia after 144 h of incubation (ANOVA F=137.936, p<0.001). Additionally, at Arctic stations Greenland Gyre and Greenland Ice-edge, no overall effect of increased CO2 on rates of DOC release, total carbon fixation or POC:DOC was observed (Poulton et al., 2016).
Overall, the observed differences in the regional response of DMSP and DMS to carbonate chemistry manipulation could not be attributed to any other measured factor that varied systematically between temperate and polar waters. These include ambient nutrient concentrations, which varied considerably but where direct manipulation had no influence on the response, and initial community structure, which was not a significant predictor of the phytoplankton response (Richier et al., 2018).
4.2 Influence of community cell-size composition on DMS response
It has been proposed that variability in the concentrations of carbonate species (e.g. pCO2, , ) experienced by phytoplankton is related to cell size, such that smaller-celled taxa (<10 µm) with a reduced diffusive boundary layer are naturally exposed to relatively less variability compared to larger cells (Flynn et al., 2012). Thus, short-term and rapid changes in carbonate chemistry, such as the kind imposed during our microcosm experiments, may have a disproportionate effect on the physiology and growth of smaller-celled species. Larger cells may be better able to cope with variability as normal cellular metabolism results in significant cell surface changes in carbonate chemistry parameters (Richier et al., 2014). Indeed, the marked response in DMS concentrations to short-term OA in temperate waters has been attributed to this enhanced sensitivity of small phytoplankton (Hopkins and Archer, 2014). Was the lack of DMS response to OA in polar waters therefore a result of the target communities being dominated by larger-celled, less carbonate-sensitive species?
Size-fractionated Chl a measurements give an indication of the relative contribution of large and small phytoplankton cells to the community. For experiments in temperate waters, the mean ratio of >10 µm Chl a to total Chl a (hereafter “”) of 0.32±0.08 was lower than the ratio for polar stations of 0.54±0.13 (Table 2). Although the difference was not statistically significant, this might imply a tendency towards communities dominated by larger cells in the polar oceans, which may partially explain the apparent lack of DMS response to elevated CO2. However, this is not a consistent explanation for the observed responses. For example, the Arctic Barents Sea station had the lowest observed “” of 0.04±0.01, suggesting a community comprised almost entirely of <10 µm cells, yet the response to short-term OA differed from the response seen in temperate waters. No significant CO2 effects on DMS or DMSP concentrations or production rates were observed at this station, whilst total Chl a significantly increased under the highest CO2 treatments after 96 h (PERMANOVA F=33.239, p<0.001). Thus, our cell size theory does not hold for all polar waters, suggesting that regardless of the dominant cell size, polar communities are more resilient to OA. In the following section, we explore the causes of this apparent insensitivity to OA in terms of the environmental conditions to which the communities have presumably adapted.
4.3 Adaptation to a variable carbonate chemistry environment
Given that DMS production by polar phytoplankton communities appeared to be insensitive to experimental OA compared to significant sensitivity in temperate communities, we hypothesize that polar communities are adapted to greater natural variability in carbonate chemistry over spatial and seasonal scales. This greater variability is partly the result of the lower buffering capacity (Revelle factor) of polar waters compared to lower-latitude waters and partly due to specific processes that occur in the polar regions that strongly alter DIC concentrations (e.g. sea-ice formation and melt, enhanced CO2 dissolution into cold polar waters, upwelling of CO2-rich water). Therefore, not only are polar plankton communities subject to geophysical processes that strongly alter in situ carbonate chemistry on both spatial and seasonal scales, but such changes are also accompanied by larger pH changes than would occur in more strongly buffered temperate waters. Therefore, polar surface ocean communities are perhaps more likely to experience fluctuations between high pH and low pH over relatively smaller timescales or space scales (Tynan et al., 2016). Thus, below, we discuss our findings in the context of the spatial pH variability we observed for each cruise track and explore some of the processes that drive this variability in polar waters. Information on the pH variability at each sampling station is not available, so we cannot be certain of the exact carbonate chemistry variability to which each of the sampled communities may have been exposed and adapted. However, we can consider the overall variability in carbonate chemistry over the spatial scales of the cruise tracks to demonstrate the characteristics of each study area.
The polar waters sampled during our study were characterized by pronounced gradients in carbonate chemistry over relatively small spatial scales. In underway samples taken along each cruise track (Arctic Ocean 3500 nm, Southern Ocean 4000 nm), pH varied by 0.45 units (8.00–8.45) in the Arctic and 0.40 units (8.30–7.90) in the Southern Ocean (Tynan et al., 2016). In some cases this range in variability was seen over relatively small distances: Figure 4 in Tynan et al. (2016) shows that pH fluctuated between 8.45 and 8.0 over a distance of 80–160 km in the sea-ice influenced Fram Strait. By comparison, pH varied by a total of 0.2 units (8.22–8.02) in underway samples from the north-western European shelf sea cruise (Rérolle et al., 2014). The observed horizontal gradients in polar waters were driven by different physical and biogeochemical processes in each ocean. In the Arctic Ocean, this variability in carbonate chemistry was partly driven by physical processes that controlled water mass composition, temperate and salinity, particularly in areas such as the Fram Strait and Greenland Sea. Along the ice edge and into the Barents Sea, biological processes exerted a strong control, as abundant iron resulted in high chlorophyll concentrations, low DIC and elevated pH. By contrast, variations in temperature and salinity had only a small influence on carbonate chemistry in the Southern Ocean in areas with iron limitation, and larger changes were driven by a combination of calcification, advection and upwelling. Where iron was replete, e.g. near South Georgia, biological DIC drawdown had a large impact on carbonate chemistry (Tynan et al., 2016). A further set of processes was in play in sea-ice influenced regions. At the Arctic ice edge, abundant iron drove strong bloom development along the ice edge, whilst sea-ice retreat in the Southern Ocean was not always accompanied by iron release (Tynan et al., 2016).
For comparison with Arctic stations, Hagens and Middelburg (2016) report a seasonal pH variability of up to 0.25 units from a single site in the open ocean surface waters in the Iceland Sea, whilst Kapsenberg et al. (2015) report an annual variability of 0.3–0.4 units in the McMurdo Sound, Antarctica. This implies that both open ocean and sea-ice influenced polar waters experience large variations in carbonate chemistry over seasonal cycles. By contrast, monthly averaged surface pCO2 data collected from station L4 in the western English Channel over the period 2007–2011 provide an example of typical carbonate chemistry dynamics in north-western European shelf sea waters. Over this period, pH had an annual range of 0.15 units (8.05–8.20), accompanied by a range in pCO2 of 302–412 µatm (Kitidis et al., 2012).
The sea-ice environment in particular is characterized by strong spatial and seasonal variability in carbonate chemistry. Sea ice is inhabited by a specialized microbial community with a complex set of metabolic and physiological adaptations allowing these organisms to withstand wide fluctuations in pH up to as high as 9.9 in brine channels to as low as 7.5 in the under-ice water (Thomas and Dieckmann, 2002; Rysgaard et al., 2012; Thoisen et al., 2015). The open waters associated with the ice edge also experience strong gradients in pH and other carbonate chemistry parameters. This can be attributed to two processes: (1) the strong seasonal drawdown of DIC due to rapid biological uptake by phytoplankton blooms at the productive ice edge which drives up pH. On the Arctic cruise, increases of up to 0.33 pH units were attributed to such processes in this region (Tynan et al., 2016). The effect was less dramatic in the Fe-limited and less productive Weddell Sea, with gradients in pH ranging from 8.20 to 8.10 (Tynan et al., 2016). (2) The drawdown of DIC is countered by the release and accumulation of respired DIC under sea ice due to the degradation of organic matter. However, this accumulation occurs in subsurface/bottom waters, which are isolated from the productive surface mixed layer by strong physical stratification and, hence, are of less relevance to the current study.
The influence of sea ice on carbonate chemistry combined with the strong biological drawdown of DIC in polar waters may have influenced the ability of some of the communities we sampled during our study to withstand the short-term changes to carbonate chemistry they experienced within the bioassays. Two of our sampling stations were “sea-ice influenced”: Greenland ice-edge and Weddell Sea. Both were in a state of sea-ice retreat as our sampling occurred in the summer months. Sampling for the Greenland ice-edge station was performed in open, deep water, near to an area of thick sea ice, with low fluorescence but reasonable numbers of diatoms (Leakey, 2012). Similarly, the Weddell Sea station was located near the edge of thick pack ice but in an area of open water that allowed sampling to occur without hindrance by brash ice (Tarling, 2013). At both stations we saw little or no response in DMS or DMSP to experimental acidification, which may imply that the in situ communities were more or less adapted to fluctuations in pH. Our experimental OA resulted in pH decreases of between 0.4 and 0.7 units. However, it is unclear whether the communities we sampled were able to withstand the artificial pH perturbation because they were adapted to living in sea ice or whether they had adapted to cope with other fluctuations in carbonate chemistry that occur in polar waters.
In summary, this demonstrates the high variability in carbonate chemistry, including pH, which polar communities may experience relative to their temperate counterparts, and which is partly driven by the lowered buffer capacity of polar waters to changes in DIC, relative to the more well-buffered temperate waters. This may have resulted in polar communities that have adapted to and are more resilient to experimentally induced OA. Of course, it is important to recognize that these data represent only a snapshot (4–6 weeks) of a year and thus do not contain information on the range in variability over daily and seasonal cycles, timescales which might be considered most important in terms of the carbonate system variability experienced by the cells and how this drives CO2 sensitivity (Flynn et al., 2012; Richier et al., 2018). Nevertheless, this inherent carbonate chemistry variability experienced by organisms living in polar waters may equip them with the resilience to cope with both experimental and future OA.
Adaptation to such natural variability may induce the ability to resist abrupt changes within the polar biological community (Kapsenberg et al., 2015). This is manifested here as negligible impacts on rates of de novo DMSP synthesis and net DMS production in the microbial communities of the polar open oceans to short-term changes in carbonate chemistry. A number of previous studies in polar waters have reported similar findings. Phytoplankton communities were able to tolerate a pCO2 range of 84–643 µatm in ∼12 d minicosm experiments (650 L) in Antarctic coastal waters, with no effects on nanophytoplankton abundance, and enhanced abundance of picophytoplankton and prokaryotes (Davidson et al., 2016; Thomson et al., 2016). In experiments under the Arctic ice, microbial communities demonstrated the capacity to respond either by selection or physiological plasticity to elevated CO2 during short-term experiments (Monier et al., 2014). Subarctic phytoplankton populations demonstrated a high level of resilience to OA in short-term experiments, suggesting a high level of physiological plasticity that was attributed to the prevailing strong gradients in pCO2 levels experienced in the sample region (Hoppe et al., 2017). Furthermore, a more recent study describing 10 CO2 manipulation experiments in Arctic waters found that primary production was largely insensitive to OA over a large range of light and temperature levels (Hoppe et al., 2018). This supports our hypothesis that, relative to temperate communities, polar microbial communities may have a high capacity to compensate for environmental variability (Hoppe et al., 2018) and are thus already adapted to, and are able to tolerate, large variations in carbonate chemistry. Thus by performing multiple, replicated experiments over a broad geographic range, the findings of this study imply that the DMS response may be both a reflection of (i) the level of sensitivity of the community to changes in the mean state of carbonate chemistry and (ii) the regional variability in carbonate chemistry experienced by different communities. This highlights the limitations associated with simple extrapolation of results from a small number of geographically limited experiments, e.g. Six et al. (2013). Such an approach lacks a mechanistic understanding that would allow a model to capture the regional variability in response that is apparent from the microcosm experiments presented here.
4.4 Comparison to an Arctic mesocosm experiment
Experimental data clearly provide useful information on the potential future DMS response to OA, but these data become most powerful when incorporated into Earth system models (ESMs) to facilitate predictions of future climate. To date, two modelling studies have used ESMs to assess the potential climate feedback resulting from the DMS sensitivity to OA (Six et al., 2013; Schwinger et al., 2017), and both have used results from mesocosm experiments. However, the DMS responses to OA within our short-term microcosm experiments contrast with the results of most previous mesocosm experiments, and, of particular relevance to this study, an earlier Arctic mesocosm experiment (Archer et al., 2013). Whilst no response in DMS concentrations to OA was generally seen in the polar microcosm experiments discussed here, a significant decrease in DMS with increasing levels of CO2 in the earlier mesocosm study was seen. Therefore, it is useful to consider how the differences in experimental design, and other factors, between microcosms and mesocosms may result in contrasting DMS responses to OA.
The short duration of the microcosm experiments (4–7 d) allows the physiological (phenotypic) capacity of the community for changes in carbonate chemistry to be assessed. In other words, how well is the community adapted to variable carbonate chemistry and how does this influence its ability to acclimate to change? Although the mesocosm experiment considered a longer time period (4 weeks), the first few days can be compared to the microcosms. No differences in DMS or DMSP concentrations were detected for the first week of the mesocosm experiment, implying a certain level of insensitivity of DMS production to the rapid changes in carbonate chemistry. In fact, when taking all previous mesocosm experiments into consideration, differences in DMS concentrations have consistently been undetectable during the first 5–10 d, implying there is a limited short-term physiological response by the in situ communities (Hopkins et al., 2010; Avgoustidi et al., 2012; Vogt et al., 2008; Kim et al., 2010; Park et al., 2014). This is in contrast to the strong response in the temperate microcosms from the north-western European shelf (Hopkins and Archer, 2014). However, all earlier mesocosm experiments have been performed in coastal waters, which like polar waters can experience a large natural range in carbonate chemistry. In the case of coastal waters this is driven to a large extent by the influence of riverine discharge and biological activity (Fassbender et al., 2016). Thus coastal communities may also possess a higher level of adaptation to variable carbonate chemistry compared to the open ocean communities of the temperate microcosms (Fassbender et al., 2016).
The later stages of mesocosm experiments address a different set of hypotheses and are less comparable to the microcosms reported here. With time, an increase in the number of generations leads to community structure changes and taxonomic shifts, driven by selection on the standing genetic variation in response to the altered conditions. Moreover, the coastal Arctic mesocosms were enriched with nutrients after 10 d, affording relief from nutrient limitation and allowing differences between pCO2 treatments to be exposed, including a strong DMS(P) response (Archer et al., 2013; Schulz et al., 2013). During this period of increased growth and productivity, CO2 increases drove changes which reflected both the physiological and genetic potential within the community and resulted in taxonomic shifts. The resultant population structure was changed, with an increase in abundance of dinoflagellates, particularly Heterocapsa rotundata. Increases in DMSP concentrations and DMSP synthesis rates were attributed to the population shift towards dinoflagellates. The drivers of the reduced DMS concentrations were less clear, but may have been linked to reduced DMSP-lyase capacity within the dominant phytoplankton, a reduction in bacterial DMSP lysis, or an increase in bacterial DMS consumption rates (Archer et al., 2013). Again, this is comparable to all other mesocosm experiments, wherein changes to DMS concentrations can be associated with CO2-driven shifts in community structure (Hopkins et al., 2010; Avgoustidi et al., 2012; Vogt et al., 2008; Kim et al., 2010; Park et al., 2014; Webb et al., 2015). However, given the lack of further experiments of a similar location, design and duration to the Arctic mesocosm, it is unclear how representative the mesocosm result is of the general community-driven response to OA in high-latitude waters.
We did not generally see any broad-scale CO2 effects on community structure in polar waters. This can be demonstrated by a lack of significant differences in the mean ratio of >10 µm Chl a to total Chl a () between CO2 treatments, implying there were no broad changes in community composition (Table 2). South Sandwich was an exception to this, where large and significant increases in the mean ratio of were observed at 750 and 2000 µatm CO2 relative to ambient CO2 (ANOVA, F=207.144, p<0.001, df=3), demonstrating that even at the short timescale of the microcosm experiments it is possible for some changes to community composition to occur. Interestingly, this was also the only polar station that exhibited any significant effects on DMS after 96 h of incubation (Fig. 4d). However, given the lack of a similar response at 1000 µatm, it remains equivocal whether this was driven by a CO2 effect or some other factor.
In contrast to our findings, a recent single 9 d microcosm experiment (Hussherr et al., 2017) performed in Baffin Bay (Canadian Arctic) saw a linear 80 % decrease in DMS concentrations during spring bloom-like conditions. It should be noted that this response was seen over a range of pCO2 from 500 to 3000 µatm, far beyond the levels used in the present study. Nevertheless, this implies that polar DMS production may be sensitive to OA at certain times of the year, such as during the highly productive spring bloom, but less sensitive during periods of low and stable productivity, such as the summer months sampled during this study. Furthermore, a number of other studies from both the Arctic, e.g. Coello-Camba et al. (2014), Holding et al. (2015), and Thoisen et al. (2015), and the Southern Ocean, e.g. Trimborn et al. (2017), Tortell et al. (2008), and Hoppe et al. (2013), suggest that polar phytoplankton communities can demonstrate sensitivity to OA, in contrast to our findings. This emphasizes the need to gain a more detailed understanding of both the spatial and seasonal variability in the polar phytoplankton community and the associated DMS response to changing ocean acidity.
We have shown that net DMS production by summertime polar open ocean microbial communities is insensitive to OA during multiple, highly replicated short-term microcosm experiments. We provide evidence that, in contrast to temperate communities (Hopkins and Archer, 2014), the polar communities we sampled were relatively insensitive to variations in carbonate chemistry (Richier et al., 2018), manifested here as a minimal effect on net DMS production. Our findings contrast with two previous studies performed in Arctic waters (Archer et al., 2013; Hussherr et al., 2017) which showed significant decreases in DMS in response to OA. These discrepancies may be driven by differences in experimental design, variable sensitivity of microbial communities to changing carbonate chemistry between different areas, or variability in the response to OA depending on the time of year, nutrient availability, and ambient levels of growth and productivity. This serves to highlight the complex spatial and temporal variability in DMS response to OA which warrants further investigation to improve model predictions.
Our results imply that the phytoplankton communities of the temperate microcosms initially responded to the rapid increase in pCO2 via a stress-induced response, resulting in large and significant increases in DMS concentrations occurring over the shortest timescales (2 d), with a lessening of the treatment effect with an increase in incubation time (Hopkins and Archer, 2014). The dominance of short response timescales in well-buffered temperate waters may also indicate rapid acclimation of the phytoplankton populations following the initial stress response, which forced the small-sized phytoplankton beyond their range of acclimative tolerance and led to increased DMS (Richier et al., 2018; Hopkins and Archer, 2014). This supports the hypothesis that populations from higher-latitude, less well-buffered waters already possess a certain degree of acclimative tolerance to variations in the carbonate chemistry environment. Although initial community size structure was not a significant predictor of the response to high CO2, it is possible that a combination of both community composition and the natural range in variability in carbonate chemistry – as a function of buffer capacity – may influence the DMS/P response to OA over a range of timescales (Richier et al., 2018).
Our findings should be considered in the context of timescales of change (experimental vs. real-world OA) and the potential of microbial communities to adapt to a gradually changing environment. Microcosm experiments focus on the physiological response of microbial communities to short-term OA. Mesocosm experiments consider a timescale that allows the response to be driven by community composition shifts, but are not long enough in duration to incorporate an adaptive response. Neither approach is likely to accurately simulate the response to the gradual changes in surface ocean pH that will occur over the next 50–100 years, nor the resulting changes in microbial community structure and distribution. However, we hypothesize that the DMS response to OA should be considered not only in relation to experimental perturbations to carbonate chemistry, but also in relation to the magnitude of background variability in carbonate chemistry experienced by the DMS-producing organisms and communities. Our findings suggest a strong link between the DMS response to OA and background regional variability in the carbonate chemistry.
Models suggest the climate may be sensitive to changes in the spatial distribution of DMS emissions over global scales (Woodhouse et al., 2013; Menzo et al., 2018). Such changes could be driven by both physiological and adaptive responses to environmental change. Accepting the limitations of experimental approaches, our findings suggest that net DMS production from polar oceans may be resilient to OA in the context of its short-term effects on microbial communities. The oceans face a multitude of CO2-driven changes in the coming decades, including OA, warming, deoxygenation and loss of sea ice (Gattuso et al., 2015). Our study addresses only one aspect of these future ocean stressors, but contributes to our understanding of how DMS emissions from the polar oceans may alter, facilitating a better understanding of Earth's future climate.
All data can be accessed via request to the British Oceanographic Data Centre. Cruise inventories and data access information can be found via the following links – Cruise JR271: https://www.bodc.ac.uk/resources/inventories/cruise_inventory/report/11432/ (last access: 13 January 2020) and Cruise JR274: https://www.bodc.ac.uk/resources/inventories/cruise_inventory/report/11606/ (last access: 13 January 2020).
The supplement related to this article is available online at: https://doi.org/10.5194/bg-17-163-2020-supplement.
CMM, SR, FEH, PDN and SDA designed the experiments. FEH and JAS conducted the measurements, and FEH and GLC analysed the data. FEH prepared the paper with assistance and contributions from all the co-authors.
The authors declare that they have no conflict of interest.
Our work and transit in the coastal waters of Greenland, Iceland and Svalbard was granted thanks to permissions provided by the Danish, Icelandic and Norwegian diplomatic authorities. We thank the captains and crew of the RRS Discovery (cruise D366) and RRS James Clark Ross (cruises JR271 and JR274) and the technical staff of the National Marine Facilities and the British Antarctic Survey. We are grateful to Mariana Ribas-Ribas and Eithne Tynan for carbonate chemistry data, Elaine Mitchell and Clement Georges for flow cytometry data, and Mariana Ribas-Ribas and Rob Thomas (BODC) for data management.
This research has been supported by the Natural Environment Research Council (NERC) (grant no. NE/H017259/1 to Philip D. Nightingale, grant no. NE/H017348/1 to C. Mark Moore), the UK Department for Environment, Food and Rural Affairs (Defra), the UK Department of Energy and Climate Change (DECC), and the National Science Foundation, United States (grant no. NSF OCE-1316133 to Stephen D. Archer).
This paper was edited by Katja Fennel and reviewed by three anonymous referees.
Archer, S. D., Kimmance, S. A., Stephens, J. A., Hopkins, F. E., Bellerby, R. G. J., Schulz, K. G., Piontek, J., and Engel, A.: Contrasting responses of DMS and DMSP to ocean acidification in Arctic waters, Biogeosciences, 10, 1893–1908, https://doi.org/10.5194/bg-10-1893-2013, 2013.
Avgoustidi, V., Nightingale, P. D., Joint, I. R., Steinke, M., Turner, S. M., Hopkins, F. E., and Liss, P. S.: Decreased marine dimethyl sulfide production under elevated CO2 levels in mesocosm and in vitro studies, Environ. Chem., 9, 399–404, 2012.
Bach, L. T., Boxhammer, T., Larsen, A., Hildebrandt, N., Schulz, K. G., and Riebesell, U.: Influence of plankton community structure on the sinking velocity of marine aggregates, Global Biogeochem. Cycles, 30, 1145–1165, 2016.
Bach, L. T., Alvarez-Fernandez, S., Hornick, T., Stuhr, A., and Riebesell, U.: Simulated ocean acidification reveals winners and losers in coastal phytoplankton, PloS one, 12, e0188198, https://doi.org/10.1371/journal.pone.0188198, 2017.
Bigg, E. K. and Leck, C.: Properties of the aerosol over the central Arctic Ocean, J. Geophys. Res.-Atmos., 106, 32101–32109, 2001.
Brussaard, C. P. D., Noordeloos, A. A. M., Witte, H., Collenteur, M. C. J., Schulz, K., Ludwig, A., and Riebesell, U.: Arctic microbial community dynamics influenced by elevated CO2 levels, Biogeosciences, 10, 719–731, https://doi.org/10.5194/bg-10-719-2013, 2013.
Carpenter, L. J., Archer, S. D., and Beale, R.: Ocean-atmosphere trace gas exchange, Chem. Soc. Rev., 41, 6473–6506, 2012.
Chang, R. Y. W., Sjostedt, S. J., Pierce, J. R., Papakyriakou, T. N., Scarratt, M. G., Michaud, S., Levasseur, M., Leaitch, W. R., and Abbatt, J. P.: Relating atmospheric and oceanic DMS levels to particle nucleation events in the Canadian Arctic, J. Geophys. Res.-Atmos., 116, https://doi.org/10.1029/2011JD015926, 2011.
Charlson, R. J., Lovelock, J. E., Andreae, M. O., and Warren, S. G.: Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate, Nature, 326, 655–661, 1987.
Chen, T. and Jang, M.: Secondary organic aerosol formation from photooxidation of a mixture of dimethyl sulfide and isoprene, Atmos. Environ., 46, 271–278, 2012.
Coello-Camba, A., Agustí, S., Holding, J., Arrieta, J. M., and Duarte, C. M.: Interactive effect of temperature and CO2 increase in Arctic phytoplankton, Front. Marine Sci., 1, 49, https://doi.org/10.3389/fmars.2014.00049, 2014.
Crawfurd, K. J., Alvarez-Fernandez, S., Mojica, K. D. A., Riebesell, U., and Brussaard, C. P. D.: Alterations in microbial community composition with increasing fCO2: a mesocosm study in the eastern Baltic Sea, Biogeosciences, 14, 3831–3849, https://doi.org/10.5194/bg-14-3831-2017, 2017.
Davidson, A. T., McKinlay, J., Westwood, K., Thompson, P., van den Enden, R., de Salas, M., Wright, S., Johnson, R., and Berry, K.: Enhanced CO2 concentrations change the structure of Antarctic marine microbial communities, Mar. Ecol. Prog. Ser., 552, 93–113, 2016.
del Valle, D. A., Kieber, D. J., Toole, D. A., Bisgrove, J., and Kiene, R. P.: Dissolved DMSO production via biological and photochemical oxidation of dissolved DMS in the Ross Sea, Antarctica, Deep-Sea Res. Pt. I, 56, 166–177, https://doi.org/10.1016/j.dsr.2008.09.005, 2009.
Egleston, E. S., Sabine, C. L., and Morel, F. M. M.: Revelle revisited: Buffer factors that quantify the response of ocean chemistry to changes in DIC and alkalinity, Global Biogeochem. Cycles, 24, GB1002, https://doi.org/10.1029/2008GB003407, 2010.
Engel, A., Zondervan, I., Aerts, K., Beaufort, L., Benthien, A., Chou, L., Delille, B., Gattuso, J.-P., Harlay, J., Heeman, C., Hoffman, L., Jacquet, S., Nejstgaard, J., Pizay, M.-D., Rochelle-Newall, E., Schneider, U., Terbrueggen, A., and Riebesell, U.: Testing the direct effect of CO2 concentrations on a bloom of the coccolithophorid Emiliania huxleyi in mesocosm experiments, Limnol. Oceanogr., 50, 493–507, 2005.
Engel, A., Schulz, K. G., Riebesell, U., Bellerby, R., Delille, B., and Schartau, M.: Effects of CO2 on particle size distribution and phytoplankton abundance during a mesocosm bloom experiment (PeECE II), Biogeosciences, 5, 509–521, https://doi.org/10.5194/bg-5-509-2008, 2008.
Eppley, R. W.: Temperature and phytoplankton growth in the sea, Fish. Bull., 70, 1063–1085, 1972.
Fassbender, A. J., Sabine, C. L., and Feifel, K. M.: Consideration of coastal carbonate chemistry in understanding biological calcification, Geophys. Res. Lett., 43, 4467–4476, https://doi.org/10.1002/2016gl068860, 2016.
Flynn, K. J., Blackford, J. C., Baird, M. E., Raven, J. A., Clark, D. R., Beardall, J., Brownlee, C., Fabian, H., and Wheeler, G. L.: Changes in pH at the exterior surface of plankton with ocean acidification, Nat. Clim. Change, 2, 510–513, 2012.
Gabric, A. J., Qu, B., Matrai, P. A., Murphy, C., Lu, H., Lin, D. R., Qian, F., and Zhao, M.: Investigating the coupling between phytoplankton biomass, aerosol optical depth and sea-ice cover in the Greenland Sea, Dynam. Atmos. Oceans, 66, 94–109, https://doi.org/10.1016/j.dynatmoce.2014.03.001, 2014.
Galindo, V., Levasseur, M., Mundy, C. J., Gosselin, M., Scarratt, M., Papakyriakou, T., Stefels, J., Gale, M. A., Tremblay, J.-É., and Lizotte, M.: Contrasted sensitivity of DMSP production to high light exposure in two Arctic under-ice blooms, J. Exp. Mar. Biol. Ecol., 475, 38–48, https://doi.org/10.1016/j.jembe.2015.11.009, 2016.
Gattuso, J.-P., Lee, K., Rost, B., and Schulz, K.: Approaches and tools to manipulate the carbonate chemistry, in: Guide to Best Practices for Ocean Acidification Research and Data Reporting, edited by: Riebesell, U., Fabry, V. J., Hansson, L., and Gattuso, J. P., Pulblications Office of the European Union, Luxembourg, 263, 2010.
Gattuso, J.-P., Magnan, A., Bille, R., Cheung, W., Howes, E., Joos, F., Allemand, D., Bopp, L., Cooley, S., and Eakin, C.: Contrasting futures for ocean and society from different anthropogenic CO2 emissions scenarios, Science, 349, aac4722, https://doi.org/10.1126/science.aac4722, 2015.
Hagens, M. and Middelburg, J. J.: Attributing seasonal pH variability in surface ocean waters to governing factors, Geophys. Res. Lett., 43, 12528–12537, https://doi.org/10.1002/2016GL071719, 2016.
Hauri, C., Friedrich, T., and Timmermann, A.: Abrupt onset and prolongation of aragonite undersaturation events in the Southern Ocean, Nat. Clim. Change, 6, 172–176, https://doi.org/10.1038/nclimate2844, 2016.
Holding, J. M., Duarte, C. M., Sanz-Martin, M., Mesa, E., Arrieta, J. M., Chierici, M., Hendriks, I. E., Garcia-Corral, L. S., Regaudie-de-Gioux, A., Delgado, A., Reigstad, M., Wassmann, P., and Agusti, S.: Temperature dependence of CO2-enhanced primary production in the European Arctic Ocean, Nat. Clim. Change, 5, 1079, https://doi.org/10.1038/nclimate2768, 2015.
Hönisch, B., Ridgwell, A., Schmidt, D. N., Thomas, E., Gibbs, S. J., Sluijs, A., Zeebe, R., Kump, L., Martindale, R. C., Greene, S. E., Kiessling, W., Ries, J., Zachos, J. C., Royer, D. L., Barker, S., Marchitto, T. M., Moyer, R., Pelejero, C., Ziveri, P., Foster, G. L., and Williams, B.: The Geological Record of Ocean Acidification, Science, 335, 1058–1063, https://doi.org/10.1126/science.1208277, 2012.
Hopkins, F. E. and Archer, S. D.: Consistent increase in dimethyl sulfide (DMS) in response to high CO2 in five shipboard bioassays from contrasting NW European waters, Biogeosciences, 11, 4925–4940, https://doi.org/10.5194/bg-11-4925-2014, 2014.
Hopkins, F. E., Turner, S. M., Nightingale, P. D., Steinke, M., Bakker, D., and Liss, P. S.: Ocean acidification and marine trace gas emissions, P. Natl. Acad. Sci. USA, 107, 760–765, 2010.
Hoppe, C. J., Schuback, N., Semeniuk, D. M., Maldonado, M. T., and Rost, B.: Functional Redundancy Facilitates Resilience of Subarctic Phytoplankton Assemblages toward Ocean Acidification and High Irradiance, Front. Marine Sci., 4, 229, https://doi.org/10.3389/fmars.2017.00229, 2017.
Hoppe, C. J. M., Hassler, C. S., Payne, C. D., Tortell, P. D., Rost, B., and Trimborn, S.: Iron Limitation Modulates Ocean Acidification Effects on Southern Ocean Phytoplankton Communities, PLOS ONE, 8, e79890, https://doi.org/10.1371/journal.pone.0079890, 2013.
Hoppe, C. J. M., Wolf, K. K. E., Schuback, N., Tortell, P. D., and Rost, B.: Compensation of ocean acidification effects in Arctic phytoplankton assemblages, Nat. Clim. Change, 8, 529–533, https://doi.org/10.1038/s41558-018-0142-9, 2018.
Hussherr, R., Levasseur, M., Lizotte, M., Tremblay, J.-É., Mol, J., Thomas, H., Gosselin, M., Starr, M., Miller, L. A., Jarniková, T., Schuback, N., and Mucci, A.: Impact of ocean acidification on Arctic phytoplankton blooms and dimethyl sulfide concentration under simulated ice-free and under-ice conditions, Biogeosciences, 14, 2407–2427, https://doi.org/10.5194/bg-14-2407-2017, 2017.
Jarníková, T. and Tortell, P. D.: Towards a revised climatology of summertime dimethylsulfide concentrations and sea–air fluxes in the Southern Ocean, Environ. Chem., 13, 364–378, https://doi.org/10.1071/EN14272, 2016.
Johnson, M. T. and Bell, T. G.: Coupling between dimethylsulfide emissions and the ocean-atmosphere exchange of ammonia, Environ. Chem., 5, 259–267, https://doi.org/10.1071/EN08030, 2008.
Kapsenberg, L., Kelley, A. L., Shaw, E. C., Martz, T. R., and Hofmann, G. E.: Near-shore Antarctic pH variability has implications for the design of ocean acidification experiments, Sci. Rep.-UK, 5, 9638, https://doi.org/10.1038/srep09638, 2015.
Kiene, R. P. and Slezak, D.: Low dissolved DMSP concentrations in seawater revealed by small-volume gravity filtration and dialysis sampling, Limnol. Oceanogr. Methods, 4, 80–95, 2006.
Kim, J. M., Lee, K., Shin, K., Kang, J. H., Lee, H. W., Kim, M., Jang, P. G., and Jang, M. C.: The effect of seawater CO2 concentration on growth of a natural phytoplankton assemblage in a controlled mesocosm experiment, Limnol. Oceanogr., 51, 1629–1636, 2006.
Kim, J. M., Lee, K., Yang, E. J., Shin, K., Noh, J. H., Park, K. T., Hyun, B., Jeong, H. J., Kim, J. H., Kim, K. Y., Kim, M., Kim, H. C., Jang, P. G., and Jang, M. C.: Enhanced Production of Oceanic Dimethylsulfide Resulting from CO2-Induced Grazing Activity in a High CO2 World, Environ. Sci. Technol., 44, 8140–8143, https://doi.org/10.1021/es102028k, 2010.
Kitidis, V., Hardman-Mountford, N. J., Litt, E., Brown, I., Cummings, D., Hartman, S., Hydes, D., Fishwick, J. R., Harris, C., and Martinez-Vicente, V.: Seasonal dynamics of the carbonate system in the Western English Channel, Cont. Shelf Res., 42, 30–40, 2012.
Korhonen, H., Carslaw, K. S., Spracklen, D. V., Mann, G. W., and Woodhouse, M. T.: Influence of oceanic dimethyl sulfide emissions on cloud condensation nuclei concentrations and seasonality over the remote Southern Hemisphere oceans: A global model study, J. Geophys. Res.-Atmos., 113, D15204, https://doi.org/10.1029/2007jd009718, 2008a.
Korhonen, H., Carslaw, K. S., Spracklen, D. V., Ridley, D. A., and Ström, J.: A global model study of processes controlling aerosol size distributions in the Arctic spring and summer, J. Geophys. Res., 113, D08211, https://doi.org/10.1029/2007JD009114, 2008b.
Lana, A., Bell, T. G., Simó, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cycles, 25, GB1004, https://doi.org/10.1029/2010GB003850, 2011.
Leaitch, W. R., Sharma, S., Huang, L., Toom-Sauntry, D., Chivulescu, A., Macdonald, A. M., von Salzen, K., Pierce, J. R., Bertram, A. K., and Schroder, J. C.: Dimethyl sulfide control of the clean summertime Arctic aerosol and cloud, Elementa: Science of the Anthropocene, 1, 000017, https://doi.org/10.12952/journal.elementa.000017, 2013.
Leakey, R.: Effect of Ocean Acidification on Arctic Surface Ocean Biology, Biogeochemistry and Climate, British Oceanographic Data Centre, Liverpool, UK, 2012.
Levasseur, M.: Impact of Arctic meltdown on the microbial cycling of sulphur, Nat. Geosci., 6, 691–700, 2013.
Lewis, E. and Wallace, D. W. R.: Program Developed for CO2 System Calculations, Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge, Tennessee, 1998.
McCoy, D. T., Burrows, S. M., Wood, R., Grosvenor, D. P., Elliott, S. M., Ma, P.-L., Rasch, P. J., and Hartmann, D. L.: Natural aerosols explain seasonal and spatial patterns of Southern Ocean cloud albedo, Sci. Adv., 1, e1500157, https://doi.org/10.1126/sciadv.1500157, 2015.
McNeil, B. I. and Matear, R. J.: Southern Ocean acidification: A tipping point at 450-ppm atmospheric CO2, P. Natl. Acad. Sci. USA, 105, 18860–18864, 2008.
Menzo, Z., Elliott, S., Hartin, C., Hoffman, F., and Wang, S.: Climate change impacts on natural sulfur production: Ocean acidification and community shifts, Atmosphere, 9, 167, https://doi.org/10.3390/atmos9050167, 2018.
Monier, A., Findlay, H. S., Charvet, S., and Lovejoy, C.: Late winter under ice pelagic microbial communities in the high Arctic Ocean and the impact of short-term exposure to elevated CO2 levels, Front. Microbiol., 5, 490, https://doi.org/10.3389/fmicb.2014.00490, 2014.
Orr, J. C., Fabry, V. J., Aumont, O., Bopp, L., Doney, S. C., Feely, R. A., Gnanadesikan, A., Gruber, N., Ishida, A., and Joos, F.: Anthropogenic ocean acidification over the twenty-first century and its impact on calcifying organisms, Nature, 437, 681–686, 2005.
Park, K.-T., Lee, K., Shin, K., Yang, E. J., Hyun, B., Kim, J.-M., Noh, J. H., Kim, M., Kong, B., Choi, D. H., Choi, S.-J., Jang, P.-G., and Jeong, H. J.: Direct Linkage between Dimethyl Sulfide Production and Microzooplankton Grazing, Resulting from Prey Composition Change under High Partial Pressure of Carbon Dioxide Conditions, Environ. Sci. Technol., 48, 4750–4756, https://doi.org/10.1021/es403351h, 2014.
Poulton, A. J., Daniels, C. J., Esposito, M., Humphreys, M. P., Mitchell, E., Ribas-Ribas, M., Russell, B. C., Stinchcombe, M. C., Tynan, E., and Richier, S.: Production of dissolved organic carbon by Arctic plankton communities: Responses to elevated carbon dioxide and the availability of light and nutrients, Deep-Sea Res. Pt. II, 127, 60–74, https://doi.org/10.1016/j.dsr2.2016.01.002, 2016.
Raven, J., Caldeira, K., Elderfield, H., Hoegh-Guldberg, O., Liss, P., Riebesell, U., Shepherd, J., Turley, C., and Watson, A.: Ocean acidification due to increasing atmospheric carbon dioxide, The Royal Society, Policy Document 12/05, London, 2005.
Rempillo, O., Seguin, A. M., Norman, A. L., Scarratt, M., Michaud, S., Chang, R., Sjostedt, S., Abbatt, J., Else, B., and Papakyriakou, T.: Dimethyl sulfide air-sea fluxes and biogenic sulfur as a source of new aerosols in the Arctic fall, J. Geophys. Res.-Atmos., 116, D00S04, https://doi.org/10.1029/2011JD016336, 2011.
Rérolle, V. M. C., Ribas-Ribas, M., Kitidis, V., Brown, I., Bakker, D. C. E., Lee, G. A., Shi, T., Mowlem, M. C., and Achterberg, E. P.: Controls on pH in surface waters of northwestern European shelf seas, Biogeosciences Discuss., 11, 943–974, https://doi.org/10.5194/bgd-11-943-2014, 2014.
Revelle, R. and Suess, H. E.: Carbon Dioxide Exchange Between Atmosphere and Ocean and the Question of an Increase of Atmospheric CO2 during the Past Decades, Tellus A, 9, 18–27, 1957.
Richier, S., Achterberg, E. P., Dumousseaud, C., Poulton, A. J., Suggett, D. J., Tyrrell, T., Zubkov, M. V., and Moore, C. M.: Phytoplankton responses and associated carbon cycling during shipboard carbonate chemistry manipulation experiments conducted around Northwest European shelf seas, Biogeosciences, 11, 4733–4752, https://doi.org/10.5194/bg-11-4733-2014, 2014.
Richier, S., Achterberg, E. P., Humphreys, M. P., Poulton, A. J., Suggett, D. J., Tyrrell, T., and Moore, C. M.: Geographical CO2 sensitivity of phytoplankton correlates with ocean buffer capacity, Global Change Biol., 24, 4438–4452, https://doi.org/10.1111/gcb.14324, 2018.
Riebesell, U., Czerny, J., von Bröckel, K., Boxhammer, T., Büdenbender, J., Deckelnick, M., Fischer, M., Hoffmann, D., Krug, S. A., Lentz, U., Ludwig, A., Muche, R., and Schulz, K. G.: Technical Note: A mobile sea-going mesocosm system – new opportunities for ocean change research, Biogeosciences, 10, 1835–1847, https://doi.org/10.5194/bg-10-1835-2013, 2013a.
Riebesell, U., Gattuso, J.-P., Thingstad, T. F., and Middelburg, J. J.: Preface: “Arctic ocean acidification: pelagic ecosystem and biogeochemicalresponses during a mesocosm study”, Biogeosciences, 10, 5619–5626, https://doi.org/10.5194/bg-10-5619-2013, 2013b.
Rysgaard, S., Glud, R. N., Lennert, K., Cooper, M., Halden, N., Leakey, R. J. G., Hawthorne, F. C., and Barber, D.: Ikaite crystals in melting sea ice – implications for pCO2 and pH levels in Arctic surface waters, The Cryosphere, 6, 901–908, https://doi.org/10.5194/tc-6-901-2012, 2012.
Sabine, C. L., Feely, R. A., Gruber, N., Key, R. M., Lee, K., Bullister, J. L., Wanninkhof, R., Wong, C. S., Wallace, D. W. R., Tilbrook, B., Millero, F. J., Peng, T.-H., Kozyr, A., Ono, T., and Rios, A. F.: The oceanic sink for anthropogenic CO2, Science, 305, 367–371, 2004.
Schoemann, V., Becquevort, S., Stefels, J., Rousseau, V., and Lancelot, C.: Phaeocystis blooms in the global ocean and their controlling mechanisms: a review, J. Sea Res., 53, 43–66, 2005.
Schulz, K. G., Riebesell, U., Bellerby, R. G. J., Biswas, H., Meyerhöfer, M., Müller, M. N., Egge, J. K., Nejstgaard, J. C., Neill, C., Wohlers, J., and Zöllner, E.: Build-up and decline of organic matter during PeECE III, Biogeosciences, 5, 707–718, https://doi.org/10.5194/bg-5-707-2008, 2008.
Schulz, K. G., Bellerby, R. G. J., Brussaard, C. P. D., Büdenbender, J., Czerny, J., Engel, A., Fischer, M., Koch-Klavsen, S., Krug, S. A., Lischka, S., Ludwig, A., Meyerhöfer, M., Nondal, G., Silyakova, A., Stuhr, A., and Riebesell, U.: Temporal biomass dynamics of an Arctic plankton bloom in response to increasing levels of atmospheric carbon dioxide, Biogeosciences, 10, 161–180, https://doi.org/10.5194/bg-10-161-2013, 2013.
Schwinger, J., Tjiputra, J., Goris, N., Six, K. D., Kirkevåg, A., Seland, Ø., Heinze, C., and Ilyina, T.: Amplification of global warming through pH dependence of DMS production simulated with a fully coupled Earth system model, Biogeosciences, 14, 3633–3648, https://doi.org/10.5194/bg-14-3633-2017, 2017.
Sharma, S., Chan, E., Ishizawa, M., Toom-Sauntry, D., Gong, S., Li, S., Tarasick, D., Leaitch, W., Norman, A., and Quinn, P.: Influence of transport and ocean ice extent on biogenic aerosol sulfur in the Arctic atmosphere, J. Geophys. Res.-Atmos., 117, D12209, https://doi.org/10.1029/2011JD017074, 2012.
Six, K. D., Kloster, S., Ilyina, T., Archer, S. D., Zhang, K., and Maier-Reimer, E.: Global warming amplified by reduced sulphur fluxes as a result of ocean acidification, Nat. Clim. Change, 3, 975–978, 2013.
Stefels, J.: Physiological aspects of the production and conversion of DMSP in marine algae and higher plants, J. Sea Res., 43, 183–197, 2000.
Stefels, J., Dacey, J. W., and Elzenga, J. T. M.: In vivo DMSP‐biosynthesis measurements using stable‐isotope incorporation and proton‐transfer‐reaction mass spectrometry (PTR‐MS), Limnol. Oceanogr.-Methods, 7, 595–611, 2009.
Steinacher, M., Joos, F., Frölicher, T. L., Plattner, G.-K., and Doney, S. C.: Imminent ocean acidification in the Arctic projected with the NCAR global coupled carbon cycle-climate model, Biogeosciences, 6, 515–533, https://doi.org/10.5194/bg-6-515-2009, 2009.
Stillman, J. H. and Paganini, A. W.: Biochemical adaptation to ocean acidification, J. Exp. Biol., 218, 1946–1955, https://doi.org/10.1242/jeb.115584, 2015.
Sunda, W., Kieber, D. J., Kiene, R. P., and Huntsman, S.: An antioxidant function for DMSP and DMS in marine algae, Nature, 418, 317–320, 2002.
Tarling, G.: Sea Surface Ocean Acidification Consortium Cruise to the Southern Ocean, British Oceanographic Data Centre, Liverpool, UK, 2013.
Thoisen, C., Riisgaard, K., Lundholm, N., Nielsen, T. G., and Hansen, P. J.: Effect of acidification on an Arctic phytoplankton community from Disko Bay, West Greenland, Mar. Ecol. Prog. Ser., 520, 21–34, 2015.
Thomas, D. N. and Dieckmann, G. S.: Antarctic Sea Ice – a Habitat for Extremophiles, Science, 295, 641–644, https://doi.org/10.1126/science.1063391, 2002.
Thomson, P. G., Davidson, A. T., and Maher, L.: Increasing CO2 changes community composition of pico- and nano-sized protists and prokaryotes at a coastal Antarctic site, Mar. Ecol. Prog. Ser., 554, 51–69, 2016.
Tortell, P. D., Payne, C. D., Li, Y., Trimborn, S., Rost, B., Smith, W. O., Riesselman, C., Dunbar, R. B., Sedwick, P., and DiTullio, G. R.: CO2 sensitivity of Southern Ocean phytoplankton, Geophys. Res. Lett., 35, L04605, https://doi.org/10.1029/2007GL032583, 2008.
Trimborn, S., Brenneis, T., Hoppe, C. J. M., Laglera, L. M., Norman, L., Santos-Echeandía, J., Völkner, C., Wolf-Gladrow, D., and Hassler, C. S.: Iron sources alter the response of Southern Ocean phytoplankton to ocean acidification, Mar. Ecol. Prog. Ser., 578, 35–50, 2017.
Tynan, E., Clarke, J. S., Humphreys, M. P., Ribas-Ribas, M., Esposito, M., Rérolle, V. M. C., Schlosser, C., Thorpe, S. E., Tyrrell, T., and Achterberg, E. P.: Physical and biogeochemical controls on the variability in surface pH and calcium carbonate saturation states in the Atlantic sectors of the Arctic and Southern Oceans, Deep-Sea Res. Pt. II, 127, 7–27, https://doi.org/10.1016/j.dsr2.2016.01.001, 2016.
Vogt, M., Steinke, M., Turner, S., Paulino, A., Meyerhöfer, M., Riebesell, U., LeQuéré, C., and Liss, P.: Dynamics of dimethylsulphoniopropionate and dimethylsulphide under different CO2 concentrations during a mesocosm experiment, Biogeosciences, 5, 407–419, https://doi.org/10.5194/bg-5-407-2008, 2008.
von Glasow, R. and Crutzen, P. J.: Model study of multiphase DMS oxidation with a focus on halogens, Atmos. Chem. Phys., 4, 589–608, https://doi.org/10.5194/acp-4-589-2004, 2004.
Webb, A. L., Malin, G., Hopkins, F. E., Ho, K. L., Riebesell, U., Schulz, K. G., Larsen, A., and Liss, P. S.: Ocean acidification has different effects on the production of dimethylsulfide and dimethylsulfoniopropionate measured in cultures of Emiliania huxleyi and a mesocosm study: a comparison of laboratory monocultures and community interactions, Environ. Chem., 13, 314–329, https://doi.org/10.1071/EN14268, 2015.
Webb, A. L., Leedham-Elvidge, E., Hughes, C., Hopkins, F. E., Malin, G., Bach, L. T., Schulz, K., Crawfurd, K., Brussaard, C. P. D., Stuhr, A., Riebesell, U., and Liss, P. S.: Effect of ocean acidification and elevated fCO2 on trace gas production by a Baltic Sea summer phytoplankton community, Biogeosciences, 13, 4595–4613, https://doi.org/10.5194/bg-13-4595-2016, 2016.
Woodhouse, M. T., Mann, G. W., Carslaw, K. S., and Boucher, O.: Sensitivity of cloud condensation nuclei to regional changes in dimethyl-sulphide emissions, Atmos. Chem. Phys., 13, 2723–2733, https://doi.org/10.5194/acp-13-2723-2013, 2013.