the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Apr 2023
| 05 Apr 2023
Ecological divergence of a mesocosm in an eastern boundary upwelling system assessed with multi-marker environmental DNA metabarcoding
Markus A. Min
David M. Needham
Sebastian Sudek
Nathan Kobun Truelove
Kathleen J. Pitz
Gabriela M. Chavez
Camille Poirier
Bente Gardeler
Elisabeth von der Esch
Andrea Ludwig
Ulf Riebesell
Alexandra Z. Worden
Eastern boundary upwelling systems (EBUS) contribute a disproportionate fraction of the global fish catch relative to their size and are especially susceptible to global environmental change. Here we present the evolution of communities over 50 d in an in situ mesocosm 6 km offshore of Callao, Peru, and in the nearby unenclosed coastal Pacific Ocean. The communities were monitored using multi-marker environmental DNA (eDNA) metabarcoding and flow cytometry. DNA extracted from weekly water samples were subjected to amplicon sequencing for four genetic loci: (1) the V1–V2 region of the 16S rRNA gene for photosynthetic eukaryotes (via their chloroplasts) and bacteria; (2) the V9 region of the 18S rRNA gene for exploration of eukaryotes but targeting phytoplankton; (3) cytochrome oxidase I (COI) for exploration of eukaryotic taxa but targeting invertebrates; and (4) the 12S rRNA gene, targeting vertebrates. The multi-marker approach showed a divergence of communities (from microbes to fish) between the mesocosm and the unenclosed ocean. Together with the environmental information, the genetic data furthered our mechanistic understanding of the processes that are shaping EBUS communities in a changing ocean. The unenclosed ocean experienced significant variability over the course of the 50 d experiment, with temporal shifts in community composition, but remained dominated by organisms that are characteristic of high-nutrient upwelling conditions (e.g., diatoms, copepods, anchovies). A large directional change was found in the mesocosm community. The mesocosm community that developed was characteristic of upwelling regions when upwelling relaxes and waters stratify (e.g., dinoflagellates, nanoflagellates). The selection of dinoflagellates under the salinity-driven experimentally stratified conditions in the mesocosm, as well as the warm conditions brought about by the coastal El Niño, may be an indication of how EBUS will respond under the global environmental changes (i.e., increases in surface temperature and freshwater input, leading to increased stratification) forecast by the IPCC.
- Article
(3100 KB) - Full-text XML
-
Supplement
(1092 KB) - BibTeX
- EndNote




Eastern boundary upwelling systems (EBUS) are exceptionally productive marine ecosystems: they account for 5 % of total global primary production (Carr, 2001) and 20 % of marine fish production (Chavez and Messié, 2009), while occupying less than 1 % of the area of the ocean. Strong physical forcing drives productivity in these ecosystems; upwelling-favorable winds bring macro- and micronutrients from depth to the surface. Under favorable temperature and light conditions, phytoplankton bloom (Messié and Chavez, 2015), leading to increases in biomass in higher trophic levels (Chavez and Messié, 2009; Ayón et al., 2008). However, these systems are variable both physically and ecologically, making it difficult to develop mechanistic understanding of the links between ecological, biogeochemical, and physical processes. Given that these systems may be disproportionately affected by climate change (Gruber, 2011) it is key that we develop a predictive understanding of how these systems will change over time. Here we present results from a perturbation experiment geared at targeting this problem.
Observational studies provide insights into processes regulating biological production and community structure in upwelling systems, but because of the complex interplay of multiple factors, it is difficult to assess the relative contributions of drivers causing change. The EBUS literature has extensive analyses of correlative relationships in the pursuit of causative understanding (e.g., Carr and Kearns, 2003; Messié and Chavez, 2015; Patti et al., 2008). By isolating natural communities in enclosures or mesocosms, one can physically perturb the system in a controlled manner (Stewart et al., 2013; Riebesell et al., 2008; Riemann et al., 2000; Sandaa et al., 2009), providing a method for studying mechanisms driving responses of these systems to perturbations. However, contained mesocosms remove horizontal mixing processes, can modify vertical mixing, and remove top predators (fish and mammals) and thus are not exact analogues of natural marine ecosystems.
Variations in zooplankton, phytoplankton, and/or bacteria have been monitored in mesocosm experiments using a variety of sampling techniques, including nets of various mesh size, direct counts of bacteria from water samples, flow cytometry for enumeration of small phytoplankton and bacteria, or algal pigment analysis (e.g., Hitchcock et al., 2016; Suffrian et al., 2008). Environmental DNA (eDNA) metabarcoding is a complementary, rapidly evolving biomonitoring technique that can survey these communities by examining both extracellular and intracellular DNA present in environmental samples (Taberlet et al., 2012). Here we define eDNA as any DNA captured by filtering seawater through a low-porosity filter (Chavez et al., 2021), and as such it includes intact microbial cells and other small live organisms as well as material shed or produced by larger plants and animals that has not yet degraded. By targeting and amplifying a highly variable region of the genome across numerous taxonomic groups, eDNA metabarcoding allows for the simultaneous detection and identification of a diversity of taxa (Valentini et al., 2016) and has been used in a variety of aquatic settings and across a wide range of organisms, recovering greater alpha diversity than visual counts or morphological identification (Djurhuus et al., 2018; Boussarie et al., 2018). While eDNA metabarcoding has been used in mesocosms to demonstrate its effectiveness by detecting and identifying known species assemblages (Kelly et al., 2014; Evans et al., 2016), its utility to detect change across multiple trophic levels has not been demonstrated in perturbation mesocosm experiments, nor have multiple eDNA markers been used simultaneously to monitor community dynamics. By providing information about broad taxonomic groups, eDNA metabarcoding provides a holistic, community-level view of ecological changes occurring within mesocosms.
Here, we present results from an in situ mesocosm experiment that took place in austral summer 2017 in the coastal Peruvian upwelling system near Callao (Bach et al., 2020). The broad goals of the experiment (detailed in Bach et al., 2020) were to study how marine populations and biogeochemical properties change during an upwelling event; nutrient-rich water collected in the regional oxygen minimum zone (OMZ) was added to the mesocosms. The mesocosms were later modified by the injection of a salt brine solution to maintain the vertical density gradient and prevent full water column mixing. This resulted in heavily stratified mesocosms. A third, unintended perturbation occurred when seabirds began to hover over and perch on the mesocosms during the last month, further modifying biogeochemical and ecological conditions. Alongside the core physicochemical measurements to characterize environmental conditions (Bach et al., 2020), there were multiple ancillary experiments. Our team collected samples for flow cytometry and eDNA from surface waters of the nearby unenclosed Pacific Ocean and from a mesocosm over the 50 d experiment period. Four genetic loci – the V1–V2 region of the 16S rRNA gene (Sudek et al., 2015; Giovannoni et al., 1990), the V9 region of the 18S rRNA gene (Amaral-Zettler et al., 2009; Stoeck et al., 2010; Amaral-Zettler et al., 2018), mitochondrial cytochrome oxidase I (COI) (Leray et al., 2013; Folmer et al., 1994), and the mitochondrial 12S rRNA gene (Miya et al., 2015) – were evaluated, capturing a diversity of bacterial, phytoplankton, zooplankton, and vertebrate populations, respectively.
Using this multi-marker approach, we detected the ecological divergence of the mesocosm relative to the nearby highly variable and dynamic unenclosed ocean. The mesocosm communities evolved to be dominated by taxa typical of stratified conditions, whereas the unenclosed ocean retained a community shaped by high nutrients and intermittent upwelling. The impact of resting seabirds was detected via an “orni-eutrophication” driven phytoplankton bloom (Bach et al., 2020) and the appearance of DNA of fish, seabirds, and bacteria typical of host-association of animal microbiomes (Saccharibacteria) (Jaffe et al., 2021). This study reveals a clear community shift driven by an experimental manipulation that simulated upwelling and subsequent stratification, as well as the impact of external inputs to the experimental system, and demonstrates that eDNA metabarcoding is a powerful tool for detecting community-level changes over time.
2.1 Mesocosm deployment and manipulations
On 22 February 2017, eight “Kiel Off-Shore Mesocosms for Ocean Simulations” (KOSMOS) (Riebesell et al., 2013) were deployed just north of Isla San Lorenzo, 6 km off the Peruvian coastline (Fig. 1). Each mesocosm consisted of a cylindrical 18.7 m long polyurethane bag (2 m diameter, 54.4 ± 1.3 m3 volume) suspended in an 8 m tall flotation frame (Bach et al., 2020). After allowing water exchange for 3 d through nets (mesh size 3 mm) at the top and bottom of each mesocosm, the water mass inside each of the mesocosms was isolated from the surrounding water by attaching a conical sediment trap to the lower end of the bags and pulling the upper ends of the bags ∼ 1.5 m above the surface. The enclosing of the mesocosms marked the start (day 0) of the 50 d experiment. Sampling of chl a and physicochemical and biogeochemical conditions was performed on all eight mesocosms as well as in the nearby coastal ocean within a few meters of the mesocosms. The eDNA and flow cytometry samples were collected from all mesocosms, but due to limited resources, the samples that were analyzed were only from mesocosm M1 and the nearby unenclosed coastal Pacific Ocean, where samples were taken within a few meters of the mesocosm frame. These two collections and their analysis are referred to as mesocosm and Pacific hereafter.
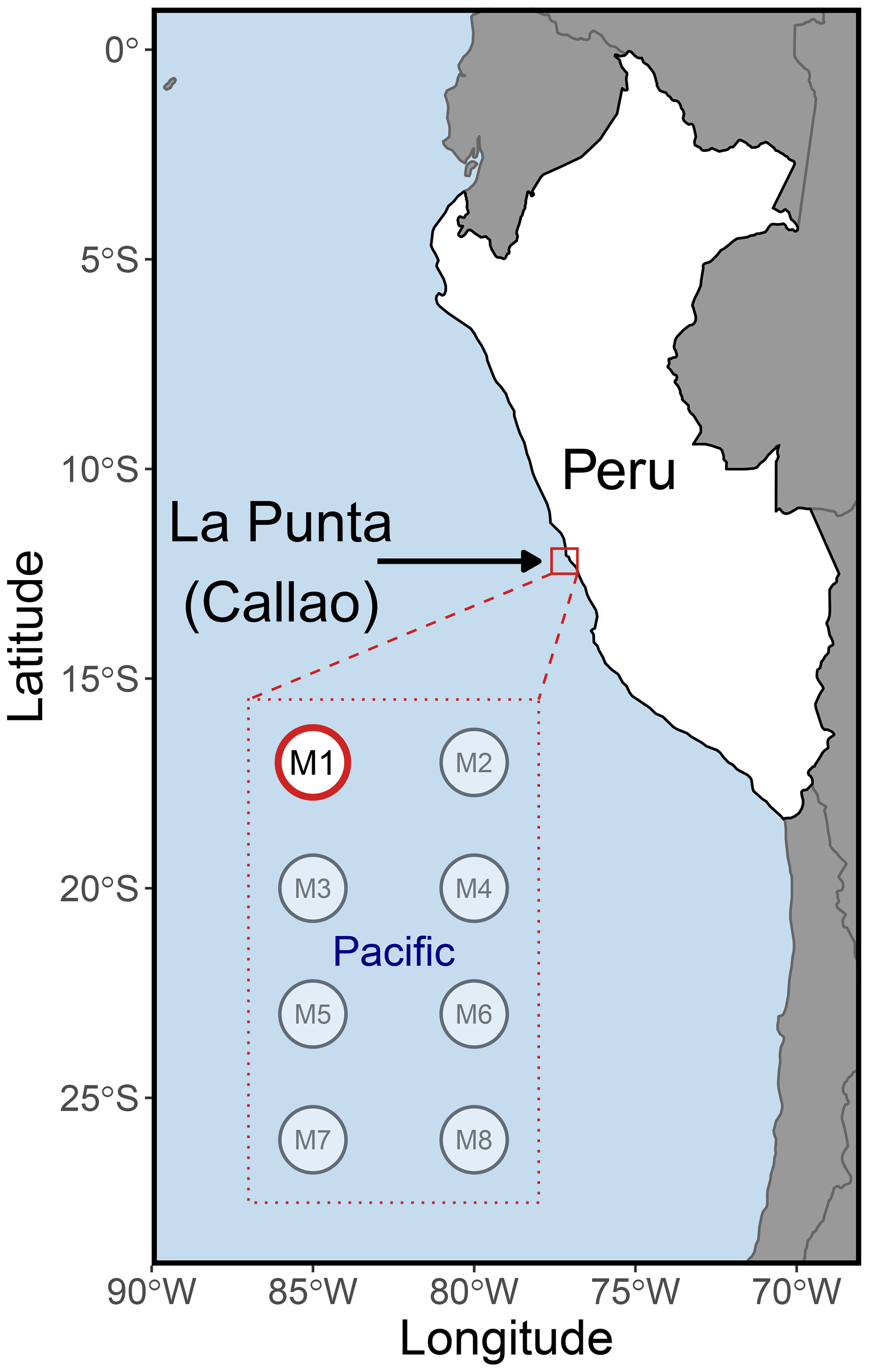
Figure 1The KOSMOS 2017 study site. The eight mesocosms (M1–M8) were deployed 6 km offshore of La Punta (Callao), just north of Isla San Lorenzo (12.0555∘ S; 77.2348∘ W). All mesocosms were sampled for chl a, various biogeochemical variables, and multiple physicochemical parameters (Bach et al., 2020). Mesocosm M1 (highlighted in red) was also analyzed for DNA and flow cytometry, as was the nearby unenclosed Pacific Ocean. Please note that the square marking the study site is not true to scale.
Over the course of the experiment, three primary intentional manipulations took place in all eight mesocosms: OMZ water addition, salt additions to control stratification, and additions of organisms. On days 11 and 12 of the experiment, a total of ∼ 20 m3 of OMZ water collected at a depth of 30 m was exchanged with the water enclosed in M1; this water had been collected on day 5 from the OMZ located at station 1 (12.028323∘ S; 77.223603∘ W) of the IMARPE (Instituto del Mar del Perú) time-series transect (Graco et al., 2017) using deep water collectors (Taucher et al., 2017). The OMZ water was injected to a depth of 14–17 m and was designed to simulate upwelling of water from the OMZ. In order to keep the mesocosm stratified and thus preserve the low O2 bottom layer from the injected OMZ water, a NaCl brine solution was injected evenly into the bottom layers of the mesocosm on days 13 (69 L at a depth range of 10–17 m) and 33 (46 L at a depth range of 12.5–17 m) of the experiment. Some of the research questions from this campaign concerned the responses of endemic organisms to the experimental conditions in the mesocosms, and therefore two endemic organisms, larvae of the Peruvian scallop (Argopecten purpuratus) and eggs of the fine flounder (Paralichthys adspersus) were added to all mesocosms. On day 14, Peruvian scallop larvae was added in concentrations of ∼ 10 000 individuals m−3, and on day 31 fine flounder eggs were added in concentrations of ∼ 90 individuals m−3. However, few scallop larvae and no fish larvae were detected during subsequent sampling via vertical tows of an Apstein net of mesh size 100 µm (unpublished data), and fine flounder DNA was only detected in the two samples directly following the addition, indicating that they either degraded or sank out of the surface layer.
2.2 Sample collection
Salinity, temperature, and chl a fluorescence were measured with vertical casts of a CTD60M sensor system (Sea and Sun Technologies) in all mesocosms and the Pacific. Samples for inorganic nutrients were collected using a 5 L “integrating water sampler” (IWS) (Hydro-Bios Kiel) that evenly collected water for two separate depth ranges, the surface and bottom waters. However, the inorganic nutrient, chl a, temperature, and salinity results presented in the first column of Fig. 2 were calculated by averaging the IWS-collected data over the two depth ranges. After transport back to an onshore laboratory, nutrient samples were filtered (0.45 µm filter, Sterivex, Merck) and analyzed using an autosampler (XY2 autosampler, SEAL Analytical) and a continuous flow analyzer (QuAAtro AutoAnalyzer, SEAL Analytical) connected to a fluorescence detector (FP-2020, JASCO). Silicic acid (Si(OH)4) was analyzed colorimetrically following the procedures by Mullin and Riley (1955). Nitrate (NO) and nitrite (NO) were quantified through the formation of a pink azo dye as established by Morris and Riley (1963). Note that other measurements were made throughout the experiment from all mesocosms as reported elsewhere (Bach et al., 2020). CTD (conductivity, temperature, and depth) casts and IWS water collections occurred every other day (except for days 1–4 and 12–18, when they were taken daily).
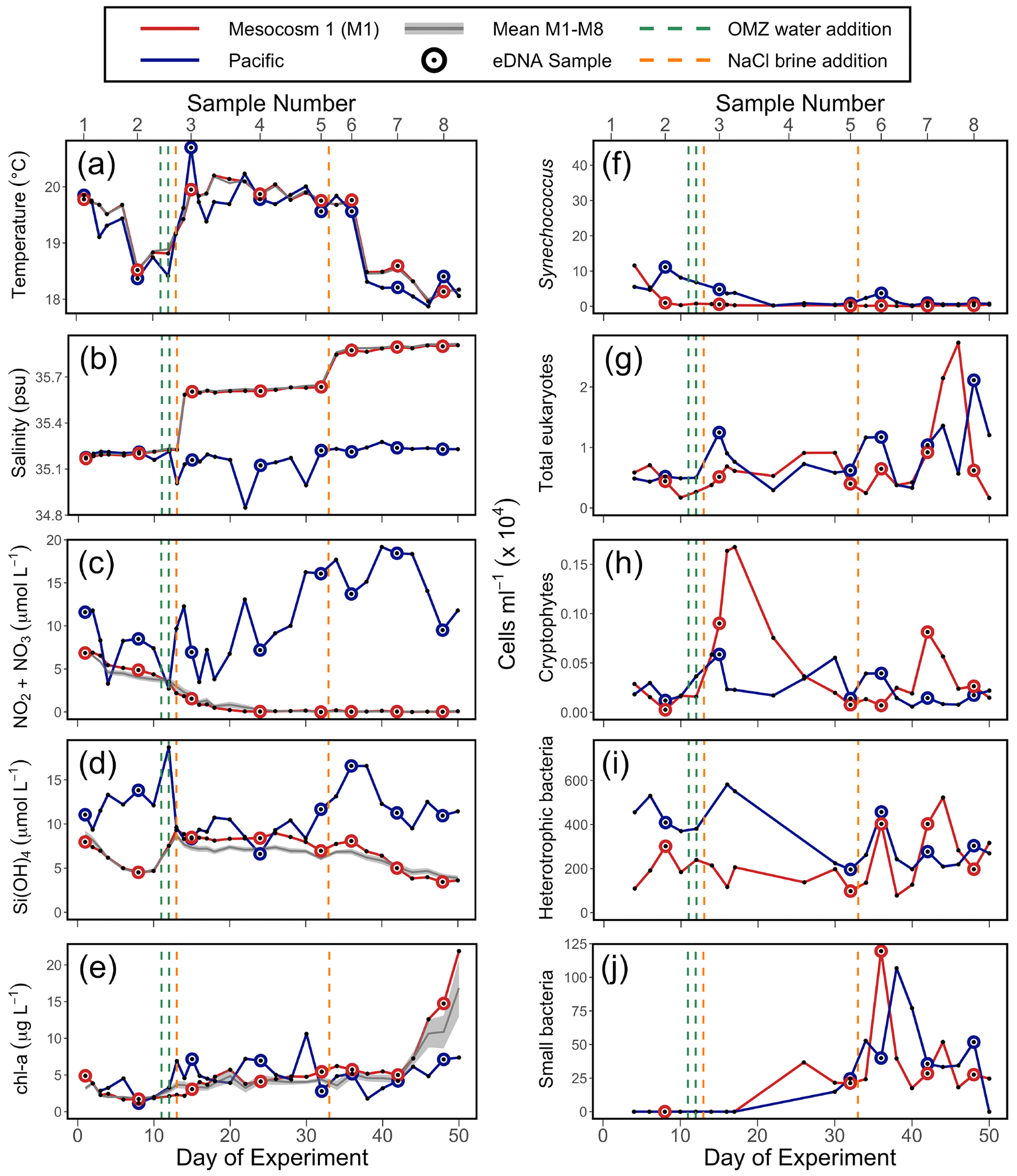
Figure 2Physicochemical conditions, inorganic nutrients, and chlorophyll a (chl a) in the mesocosms and unenclosed Pacific surface waters (a–e), with values averaged over the 0–17 m depth range. Flow cytometry results (f–j) from samples collected in the surface waters; these depths were 0–5 m on days 1 and 2, 0–10 m from day 3 to 28, and 0–12.5 m from day 29 to 50. For all panels, each individual sample is indicated. For (a), (b), (c), (d), and (e), the solid grey line represents the mean for all eight mesocosms; the shaded grey line is the mean ± SE for all eight mesocosms. The green lines indicate exchange of ∼ 20 m3 of OMZ water with water in the mesocosm (total volume ∼ 50 m3) on days 11 and 12. The orange lines indicate NaCl brine additions on days 13 (69 L at a depth range of 10–17 m) and 33 (46 L at a depth range of 12.5–17 m). (a) Temperature. (b) Salinity. (c) NONO. (d) Si(OH)4. (e) Chlorophyll a (chl a). (f) Synechococcus. (g) Photosynthetic eukaryotes. (h) Cryptophytes. (i) Heterotrophic (non-pigmented) bacteria. (j) Small bacteria (smaller than standard non-pigmented bacteria in the system).
DNA samples that were analyzed were taken in M1 and the Pacific on days 1, 8, 15, 24, 32, 36, 42, and 48. A single DNA sample was analyzed for each day. Flow cytometry samples were collected roughly every other day. Water for eDNA and flow cytometry measurements was collected using the IWS to evenly sample the upper portion of the water column; the depths sampled were 0–5 m on days 1 and 2, 0–10 m from day 3 to 28, and 0–12.5 m from day 29 to 50. These sampling depths sampled the top layer of the mesocosm, which was almost entirely above the depths at which the salt brine solution was injected. After transport back to shore, eDNA samples were collected by filtering 250 mL of water onto a 47 mm diameter 0.22 µm pore size polyvinylidene difluoride membrane filter (Millipore, USA) using a vacuum pump. All filters were flash frozen in liquid nitrogen and stored at −80 ∘C before being shipped on dry ice to California, USA, for analysis.
Field controls consisting of 250 mL of filtered reverse osmosis (RO) and MilliQ water were collected on day 3 of the experiment in order to characterize any contaminating taxa in these systems and later steps. No DNA was detected in these controls using NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA) measurements; environmental samples ranged from 19.9–139.3 ng µL−1. However, COI, 18S, and 16S rRNA PCR amplification yielded slight amplification, and PCR products were sequenced (Table S1). After normalization steps, the dominant reads in the bacterial portion of the 16S rRNA sequences consisted of Betaproteobacteria (36.2 %) that were not prominent in field samples (0.27 % mean proportion of reads in field samples). The greatest proportion of plastidial reads in the field blanks were multiple diatom amplicon sequence variants (ASVs) (66.7 % of reads), with the same ASVs composing 42.8 % of reads in the field samples. In the COI reads, most ASVs found in the field blanks were also not prominent in the field samples, with the exception of the calanoid copepod Paracalanus (12.4 % of reads in field blanks, 13.3 % of reads in field samples). In the 18S reads, the top ASVs in the field blanks were the copepods Hemicyclops thalassius (30.0 % of reads in field blanks, 3.9 % of reads in environmental samples) and Paracalanus (11.7 % of reads in field blanks, 14.6 % of reads in environmental samples). While this indicates that there may have been a low level of field-based cross-contamination in our environmental samples, which is found in most eDNA studies (and is a near inevitability when sampling in a remote location without a dedicated molecular laboratory), these reads were not removed from our analyses.
2.3 Flow cytometry
Triplicate samples of 1 mL volume each were taken and preserved with glutaraldehyde (EM grade, final concentration of 0.25 %). The samples were then incubated for 20 min in the dark and subsequently treated the same way as the eDNA samples. Photoautotrophs (photosynthetic eukaryotes, Synechococcus, and Prochlorococcus were present) and heterotrophic bacteria were enumerated on a BD Influx cell sorter (BD Biosciences, USA) equipped with a 488 nm argon laser (200 mW) and a 70 µm nozzle running with 0.2 µm pre-filtered 1X PBS (10X PBS, pH 7.4, Life Technologies). Prior to the running of each sample, fluorescent polystyrene beads (0.75 µm yellow green beads, Polysciences, Inc) were added for reference. For calculation of the total volume analyzed, the samples were weighed before and after each run. For counts of photoautotrophs, the system was triggered on forward angle light scatter (FALS), and red chlorophyll autofluorescence (692/40 nm band-pass filter) as well as orange phycoerythrin autofluorescence (572/27 nm band-pass filter) versus FALS were recorded over 8 min, running at ∼ 25 µL min−1. To count heterotrophic bacteria, the samples were stained with SYBR Green I (10 000× SYBR Green I, Thermo Fisher; final concentration of 0.5× with 15 min incubation time in the dark) and was triggered on green fluorescence (520/35 nm band-pass filter). The samples were run for 1 min. at ∼ 25 µL min−1.
Flow Cytometry Standard (FCS) files were processed in WinList 3D 9.0.1 (Verity Software House, Topsham ME, USA). Among other bacterial populations, a unique population of presumed bacteria appeared in mesocosm and coastal samples after day 24 of the study and was gated in accordance with the representative cytogram (Fig. S1).
2.4 DNA extraction
DNA from filters was extracted using the DNeasy® Blood and Tissue kit (Qiagen, Germantown, MD) following standard protocol, with some modifications that included an overnight incubation and increasing the amount of lysis buffer to completely submerge the filter during lysis (Walz et al., 2019). DNA extraction concentrations were quantified using NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA) measurements.
2.5 DNA amplification and sequencing
2.5.1 Cytochrome oxidase I (COI), 18S rRNA, and 12S rRNA
PCR reactions for COI and the 12S rRNA gene were run with Fluidigm two-step amplification protocol for each sample (COI, Closek et al., 2018b; 12S, Pitz et al., 2020), while PCR reactions for the 18S rRNA gene were run using 12 basepair Golay barcoded reverse primers (Closek et al., 2018a). For COI, the primary PCR primers used are from Leray et al., 2013, and are as follows: Fluidigm CS1 + mlCOIinfF (forward) is ACACTGACGACATGGTTCTACA GGWACWGGWTGAACWGTWTAYCCYCC, and Fluidigm CS2+HCO2198 (reverse) is TACGGTAGCAGAGACTTGGTCT TAAACTTCAGGGTGACCAAAAAATCA. For 18S, primary PCR primers used are from Amaral-Zettler et al. (2009) and are as follows: Euk1391F (forward) is AATGATACGGCGACCACCGAGATCTACAC TATCGCCGTT CG GTACACACCGCCCGTC, and EukBr (reverse) is CAAGCAGAAGACGGCATACGAGAT XXXXXXXXXXXX AGTCAGTCAG CA TGATCCTTCTGCAGGTTCACCTAC (where XXXXXXXXXXXX is a unique 12 bp barcode location; all primers listed in 5′ to 3′ direction). For 12S, primary PCR primers used are from Miya et al. (2015) and are as follows: Fluidigm CS1 + 12S MiFish_U (forward) is ACACTGACGACATGGTTCTACAGTCGGTAAAACTCGTGCCAGC and Fluidigm CS2 + 12S MiFish_U (reverse) is
TACGGTAGCAGAGACTTGGTCT
CATAGTGGGGTATCTAATCCCAGTTTG.
For each of the three markers, primary PCR amplifications were carried out in triplicate 25 µL reactions using 1 µL DNA extract, 12.5 µL AmpliTaq Gold Fast PCR master mix (Applied Biosystems), 1 µL each of forward and reverse primers (5 µM), and 9.5 µL molecular-biology-grade water. PCR reactions were run in 96-well plates with a no template control (NTC) run in triplicate for each plate. Primary COI cycling parameters were 95 ∘C for 10 min followed by 16 cycles of 94 ∘C for 10 s; 62 ∘C for 30 s; 68 ∘C for 60 s, next followed by 25 cycles of 94 ∘C for 10 s; 46 ∘C for 30 s; 68 ∘C for 60 s; and a final step of 72 ∘C for 10 min. Primary 18S rRNA cycling parameters were 95 ∘C for 10 min followed by 35 cycles of 94 ∘C for 45 s, 57 ∘C for 30 s, 68 ∘C for 90 s, and a final elongation step of 72 ∘C for 10 min. Primary 12S rRNA cycling parameters were 95 ∘C for 15 min followed by 13 cycles of 94 ∘C for 30 s; 69.5 ∘C for 30 s (changes −1.5 ∘C per cycle); 72 ∘C for 90 s, next followed by 25 cycles of 94 ∘C for 30 s; 50 ∘C for 30 s; 72 ∘C for 45 s; and a final step of 72 ∘C for 10 min.
Following PCR, the pooled PCR products for each genetic marker were run through an agarose gel to confirm the presence of target bands and inspected for degree of amplification as well as absence of any non-specific amplification. PCR products were purified and size selected using the Agencourt AMPure XP bead system (Beckman Coulter, USA). A second agarose gel was run to confirm primer removal and retention of target amplicons after purification.
Library preparation and sequencing was conducted at the Research Technology Support Facility (RTSF) Genomics Core at Michigan State University (MSU), as was secondary amplification for COI and 12S. PCR products were run through a Invitrogen SequalPrep Normalization Plate (Thermo Fisher Scientific) using the manufacturer's protocol to create pooled libraries. The pooled product was loaded on a standard MiSeq v2 flow cell and sequenced in a 2×250 bp (COI, 12S rRNA) or 2×150 bp (18S rRNA) paired end format using a v2 500-cycle MiSeq reagent cartridge. The MiSeq run was performed with a 10 % PhiX spike. Custom sequencing primers were added to appropriate wells of the reagent cartridge. Base calling was done by Illumina real time analysis (RTA) v1.18.54, and output of RTA was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v2.18.0.
2.5.2 16S rRNA
Prior to amplification, DNA was diluted to 5 ng µL−1 with TE pH 8. The V1–V2 16S rRNA gene region was amplified as previously described (Sudek et al., 2015), with 5 µL of 10× buffer, 1 U of HiFi-Taq, 1.6 mM MgSO4, 5 ng of template DNA, and 200 nM of 27F (AGRGTTYGATYMTGGCTCAG; Daims et al., 1999) and 338RPL primer (GCWGCCWCCCGTAGGWGT; Morris et al., 2002). PCR cycling parameters were 95 ∘C for 2 min, 30 cycles of 94 ∘C for 15 s, 55 ∘C for 30 s, and 68 ∘C for 1 min, followed by a final elongation at 68 ∘C for 7 min. Purification, barcoding, library preparation, and sequencing were performed at the University of Arizona with MiSeq 2×300 bp reads.
2.6 Bioinformatics
2.6.1 Cytochrome oxidase I (COI), 18S rRNA, and 12S rRNA
The resulting Illumina sequence data were analyzed through a custom shell script adapted from the banzai pipeline (https://github.com/MBARI-BOG/BOG-Banzai-Dada2-Pipeline, last access: 20 October 2020; O'donnell et al., 2016). Complete script and parameters are included in the Supplement. Within the script, primer sequences were first removed from fastq files through the program Atropos (Didion et al., 2017). Fastq files were then fed into the DADA2 program (Callahan et al., 2016). DADA2 models error on a per-Illumina run basis, controlling for read quality and picking ASV sequences that represent biological variability rather than sequencing error (Callahan et al., 2016). Within DADA2, reads were trimmed to remove low-quality regions and filtered by quality score, sequencing errors were modeled and removed, and reads were then merged and chimeric sequences removed. Taxonomy was assigned to the resulting ASV sequences through blastn searches to NCBI GenBank's non-redundant nucleotide database (nt) (Camacho et al., 2009; Agarwala et al., 2018). Blast results were filtered using MEGAN6's lowest common ancestor (LCA) algorithm (Huson et al., 2016). Only hits with ≥ 80 % sequence identity, with ≥ 100 bitscore, and whose bitscores were within the top 2 % of the highest bitscore value for each ASV were considered by MEGAN6. The MEGAN6 parameter LCA percent was from 0.80 to 0.85, depending on the marker, allowing for up to 15 %–20 % of top hits to be off target and still have the majority taxonomy assigned. This parameter value was chosen to allow for minor numbers of incorrectly annotated GenBank entries – effectively allowing for ASVs which had many high-quality hits to a taxa to still be assigned to that taxa even if there existed a high-bitscore hit to another GenBank sequence annotated to an unrelated taxa. Despite the issue that this introduces potentially assigning species-level identification to an ASV with multiple strong matches, we decided that this compromise was necessary to allow species identification with an imperfectly curated reference database. We decided this was more advantageous than the disadvantage caused by ignoring small numbers of true closely related sequences. Furthermore, post-MEGAN6 filtering was performed to ensure only contigs with a hit of ≥ 97 % sequence identity and ≥ 200 bitscore were annotated to the species level. Only contigs with a hit of ≥ 95 % sequence identity and ≥ 150 bitscore were annotated to the genus level. Annotations were elevated to the next highest taxonomic level for contigs that failed those conditions.
2.6.2 16S rRNA
Demultiplexed reads were imported into QIIME2 (Bolyen et al., 2019) and cutadapt (Martin, 2011) trim-paired was used to trim primers. Trimmed reads were denoised with DADA2 (Callahan et al., 2016) denoise-paired command, with a truncation of the forward and reverse reads to 250 and 225 bp, respectively. Resulting ASVs were classified in QIIME2 with the feature-classifier classify-consensus-blast (Camacho et al., 2009) command, with a percent identity of 0.95, maximum number of accepted hits of 5 and a consensus of 0.7 against the 99 % representative sequences of SILVA 132 (Quast et al., 2013) and both 18S and 16S rRNA gene references. A bacterial ASV table was then generated by removing mitochondrial and plastidial sequences. Plastidial sequences were then selected based on the SILVA classification and reclassified with the Phytoref database (Decelle et al., 2015) using the same settings as before except with a percent identity of 0.90. The five most abundant cyanobacterial ASVs were further analyzed by a manual blast against NCBI-nt excluding environmental sequences.
2.7 Quality control and decontamination
Following the bioinformatic pipeline, the COI, 18S rRNA, and 12S rRNA sequencing results were passed through custom R ver. 3.6.0 (R Core Team, 2019) decontamination scripts. For each plate, we first removed all singleton ASVs. Next, for each ASV that was detected in at least one of the PCR blanks on the plate, we determined the maximum number of reads of that ASV in any of the individual PCR blanks and subtracted this value from the reads of the ASV in each of the environmental samples. This was done to address cross-contamination from various sources, such as tag jumping (Schnell et al., 2015). As mentioned previously in Sect. 2.2, no decontamination steps were taken using the field blank samples. Finally, we removed all reads assigned to common terrestrial contaminants: orders Rodentia and Lagomorpha; families Hominidae, Bovidae, Felidae, and Canidae; and genera Gallus and Meleagris. These decontamination steps were not run for 16S rRNA sequences because the PCR blanks were not sequenced.
2.8 Statistical analyses
Beta diversity analyses were run separately on the five datasets (COI, 18S rRNA, 12S rRNA, 16S rRNA bacterial sequences, and 16S rRNA plastidial sequences) using the QIIME2 (Bolyen et al., 2019) DEICODE plugin. The beta diversity analyses for 12S rRNA are included in the Supplement (Fig. S2) since vertebrates were excluded from the mesocosm (water filtered through a 3 mm mesh), and vertebrate eDNA that was detected was (1) found during our first sampling, and then it decayed and disappeared at a rate consistent with experimental results (Sassoubre et al., 2016); (2) the result of the intentional addition of fish eggs; and (3) the unintentional addition from seabird faeces. Through matrix completion and robust Aitchison PCA (RPCA), DEICODE (Martino et al., 2019) is particularly well-suited to handle the sparseness inherent to sequencing data. A PERMANOVA test was also run in QIIME2 on the Aitchison distance matrix produced by DEICODE to determine the significance of the variance between M1 samples following OMZ water addition and the other samples (Pacific and first two M1 samples). Based on the results of the RPCA and PERMANOVA, which showed a clear differentiation between the M1 samples following OMZ water addition (day 15 and on) and Pacific surface water samples on PC1 for the four primary datasets analyzed for beta diversity (all but 12S rRNA), the loading scores on PC1 were used to identify ASVs that were driving the divergence between M1 and the Pacific. The PC results presented are constrained to ASVs that were consistently present throughout the experiment, as determined by ASVs that composed at least 0.1 % of the reads in 25 % of the samples. Heatmaps of relative abundances were constructed for all ASVs in all samples (Fig. S3); the criteria for inclusion of ASVs in these heatmaps was a relative abundance within the top 10 most abundant ASVs in any sample within a respective dataset and greater than 1 % of the reads in any sample.
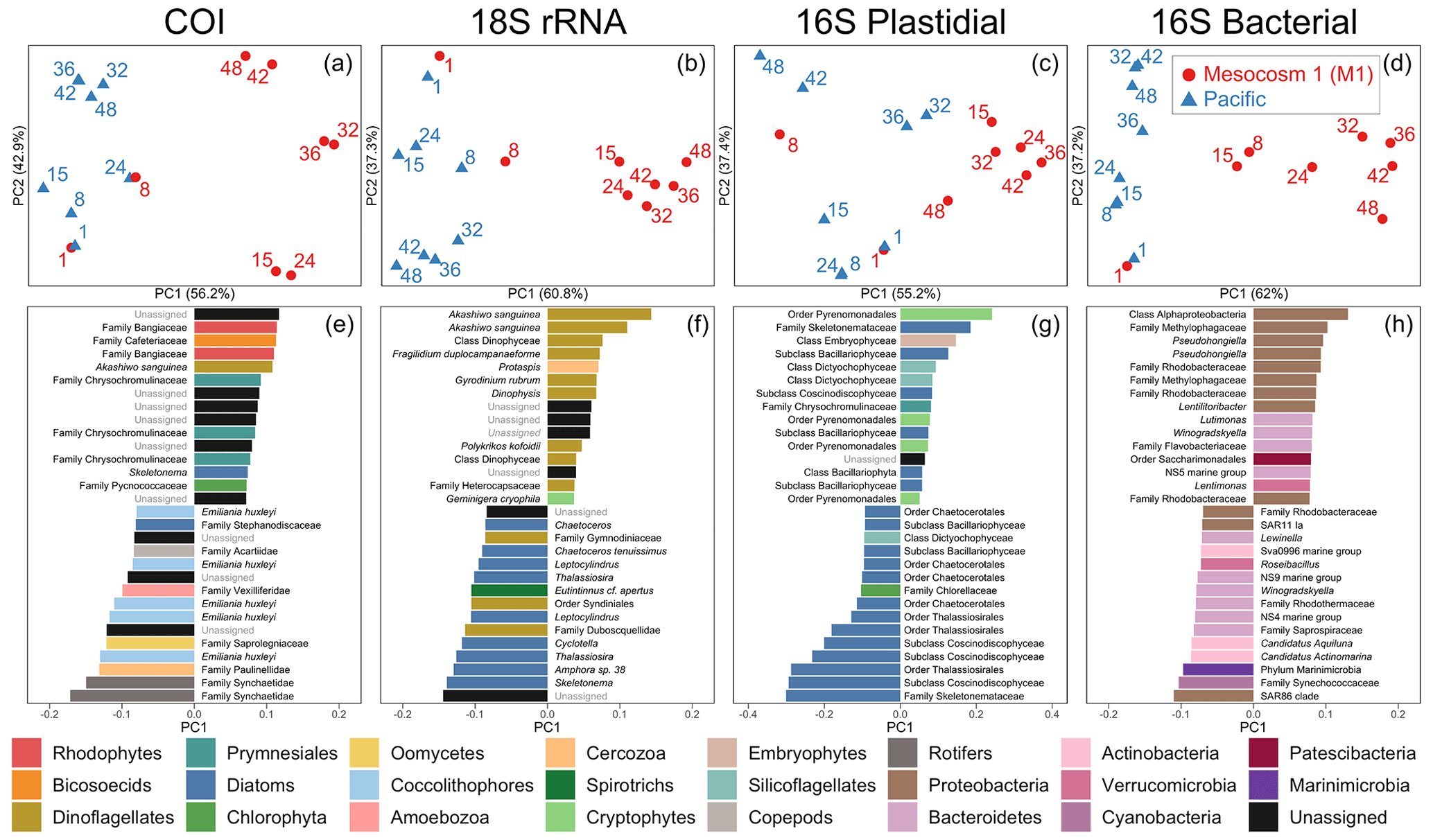
Figure 3Beta diversity analyses using DEICODE robust Aitchison PCA for (a) COI, (b) 18S rRNA, (c) V1–V2 16S rRNA plastidial ASVs, and (d) V1–V2 16S rRNA bacterial ASVs, including photosynthetic taxa. Data points represent individual samples; the number next to each data point indicates the day collected. ASV loadings on PC1 from the DEICODE robust Aitchison PCA for (e) COI, (f) 18S rRNA, (g) V1–V2 16S rRNA plastidial ASVs, and (h) V1–V2 16S rRNA bacterial ASVs. As seen in (a), (b), (c), and (d) and confirmed by a PERMANOVA test, PC1 separates the Pacific samples and pre-OMZ water addition M1 samples from the post-OMZ water addition M1 samples (days 15+), and thus positive loading scores are associated with the mesocosm, whereas negative loading scores are associated with the Pacific samples. The text adjacent to the loading scores represents the lowest taxonomy to which that ASV was annotated. The taxonomy assigned to 16S rRNA ASVs corresponds to that of the SILVA taxonomy.
3.1 Physicochemical conditions, inorganic nutrients, and chlorophyll a
The mesocosm experiment occurred during an unusual warming event described as a coastal El Niño (Garreaud, 2018; Bach et al., 2020). Sea surface temperature (SST) was 1 to 1.5 ∘C warmer than average (Herring et al., 2019) at the start of the experiment. SST fluctuated over the experiment (Fig. 2a) but returned to average values about 5 weeks into the experiment. Temperature in the mesocosms tracked that of the surrounding water (Fig. 2a). Salinity in the mesocosms began with levels similar to the open ocean and was modified via NaCl brine additions to depths below 10 m on day 13 and again on day 33 (Fig. 2b).
NO NO (NO) concentration in M1 was initially 6.9 µmol L−1 and declined steadily over time (Fig. 2c). M1 received addition of OMZ water with a NO concentration of 0.3 µmol L−1 on days 11 and 12 and reached the detection threshold of 0.2 µmol L−1 around day 20 (Fig. 2c). Conversely, NO concentrations in the Pacific at the study site were considerably higher and more variable, ranging between 2.7–19.2 µmol L−1 and reaching particularly high values during the second half of the experiment (Fig. 2c). The concentration of Si(OH)4 in M1 was initially 8.0 µmol L−1 and declined to a value of 4.7 µ mol L−1 on day 10, prior to the addition of OMZ water with a Si(OH)4 concentration of 17.4 µmol L−1. The addition of OMZ water caused Si(OH)4 in M1 to increase to 9.3 µmol L−1 on day 13, after which its concentration declined for the rest of the experiment, reaching a value of 3.6 µmol L−1 on day 50 (Fig. 2d). In the Pacific, Si(OH)4 concentrations fluctuated, ranging from a minimum of 6.6 µmol L−1 on day 24 to a maximum of 18.7 µmol L−1 on day 12 (Fig. 2d).
Chlorophyll a (chl a) concentration in M1 was initially 4.9 µg L−1 and declined until reaching a minimum of 1.7 µg L−1 on day 6. Following OMZ water addition, chl a gradually increased until approximately day 40, after which it increased rapidly. This rapid increase in chl a in the final ∼ 10 d of the experiment, which was seen across all eight mesocosms (Fig. 2e), has been attributed to orni-eutrophication by defecating seabirds (Inca terns, Larosterna inca), who found the mesocosms to be suitable resting places (Bach et al., 2020). Chl a in the Pacific was more variable and generally comparable to the range of chl a concentrations found in the mesocosms until day 40.
The observed values of temperature, salinity, NO, Si(OH)4, and chl a in M1 were very similar to the values found in all eight mesocosms (Fig. 2, with more detail in Bach et al., 2020), indicating that M1 was representative of the overall abiotic and hence biotic conditions found within the mesocosms.
3.2 eDNA overview statistics
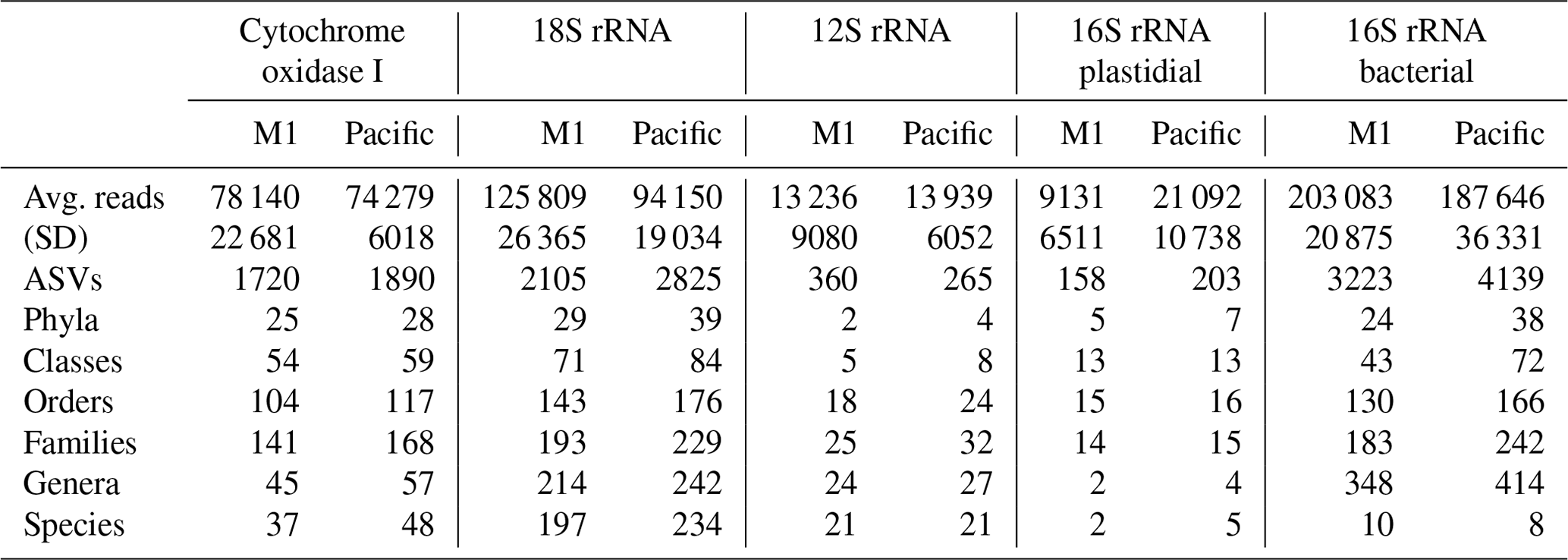
Across the five datasets, the unenclosed Pacific samples exhibited higher alpha diversity relative to mesocosm at nearly all levels of taxonomy from phyla to ASVs. Summary statistics are given in Table 1. A total of 1 219 350 COI, 1 759 671 18S rRNA, 217 398 12S rRNA, 241 783 16S rRNA plastidial, and 3 125 836 16S rRNA bacterial paired end reads passed filtering and decontamination steps. These amplicons resulted in a total of 2434 COI ASVs, 3296 18S rRNA ASVs, 470 12S rRNA ASVs, 258 16S rRNA chloroplast ASVs, and 5786 16S rRNA bacterial ASVs (Table 1). These resulting ASVs form the basis of the statistical analysis presented below.
Table 1DNA sequencing statistics. The 16 total samples were divided between 8 M1 (mesocosm) and 8 Pacific samples, with “Avg. reads” and “Stdev” reflecting the average and standard deviation (respectively) of the number of reads per sample. All other values represent the total sum across all samples. Across all five datasets, the Pacific samples consistently show a higher level of alpha diversity at all levels of taxonomy.
3.3 Results of principal component analysis
3.3.1 RPCA and PERMANOVA
The first principal component of COI, 18S rRNA, V1–V2 16S rRNA plastidial ASVs, and V1–V2 16S rRNA bacterial ASVs revealed a swift divergence between the Pacific and mesocosm communities (Fig. 3). In the COI, 18S rRNA, and 16S rRNA plastidial sequences (Fig. 3a, b, and c), this divergence is first seen on day 15, with the first eDNA sample taken after the OMZ water and salt additions. In the 16S rRNA bacterial sequences (Fig. 3d), this divergence is already seen on day 8, prior to additions. A PERMANOVA test revealed a significant difference between M1 samples following additions and the other samples, confirming the significance of the observed divergence (COI: p=0.001, F statistic = 11.78; 18S rRNA: p=0.001, F statistic = 11.76; 16S rRNA chloroplast: p=0.002, F statistic = 7.20; 16S rRNA bacterial: p=0.001, F statistic = 10.11). Following this initial divergence, the M1 samples continue to separate from the Pacific samples. The Pacific samples show a slight temporal trend on PC2 across all four datasets. As shown by the proportion of variance explained by each principal component, the degree of divergence between M1 and the Pacific is greater than the degree of change experienced by the Pacific.
3.3.2 Mesocosm-associated ASVs and taxonomy revealed by PC1 loading scores
The taxa that had the highest positive PC1 loading scores, and thus were associated with M1, included various photo-, mixo-, and heterotrophic taxa. For the COI data, this included the dinoflagellate Akashiwo sanguinea. Unassigned ASVs made up six of the other mesocosm-associated ASVs for COI, while other mesocosm-associated ASVs included two assigned to the red algae family Bangiaceae, one to the heterotrophic nanoflagellate family Cafeteriaceae, three to the haptophyte family Chrysochromulinaceae, one to the diatom Skeletonema, and one to the prasinophyte family Pycnococcaceae (Class V prasinophytes; Bachy et al., 2022).
The mesocosm-associated 18S rRNA ASVs were dominated by dinoflagellates, which were 9 of the top 15 positive loading ASVs. The six other ASVs in the top 15 mesocosm-associated ASVs included one ASV assigned to the cercozoan genus Protaspis, another ASV assigned to the cryptophyte species Geminigera cryophila, and four unassigned ASVs.
In the V1–V2 16S rRNA plastidial sequences, mesocosm-associated ASVs included four ASVs assigned to the cryptophyte order Pyrenomonadales, six diatom ASVs, an ASV assigned to the class Embryophyceae (land plants), two ASVs assigned to the silicoflagellates (class Dictyochophyceae), an ASV assigned to the family Chrysochromulinaceae, and an unassigned ASV. In the V1–V2 16S rRNA bacterial sequences, nine ASVs were assigned to the phylum Proteobacteria, four to the phylum Bacteroidetes, and one each to the phyla Patescibacteria and Verrucomicrobia.
3.3.3 Pacific ASVs and taxonomy revealed by PC1 loading scores
Pacific COI ASVs included three mesozooplankton ASVs, two assigned to the rotifer family Synchaetidae and one assigned to the calanoid copepod family Acartiidae. In the phytoplankton portion of the COI Pacific-associated ASVs, the coccolithophore Emiliania huxleyi figured prominently, comprising five of the top 15 Pacific-associated ASVs. The diatom family Stephanodiscaceae was also determined to be associated with the Pacific. Of the COI ASVs associated with the Pacific that were not mesozooplankton or phytoplankton, one ASV belonged to the amoeboid family Paulinellidae (phylum Cercozoa), one was assigned to the amoebozoan family Vexilliferidae, one was assigned to the oomycete family Saprolegniaceae, and three ASVs were unassigned.
Pacific 18S rRNA ASVs were dominated by diatom ASVs, which made up 9 of the top 15 Pacific-associated ASVs. All of these diatom ASVs were annotated to the genus- or species-level, and included the genera Skeletonema, Amphora, Thalassiosira, Cyclotella, Leptocylindrus, and Chaetoceros. Three of the top 15 Pacific ASVs were dinoflagellate ASVs, two of which were annotated to the order Syndiniales, with one of these ASVs annotated to the family Duboscquellidae within the Syndiniales; the other dinoflagellate ASV was annotated to the family Gymnodiniaceae. One other ASV was assigned to the tintinnid ciliate species Eutintinnus cf. apertus, and two other ASVs were unassigned.
The Pacific V1–V2 16S rRNA plastidial ASVs were dominated by diatoms, which represented 13 of the top 15 Pacific-associated ASVs. Of the two other Pacific-associated chloroplast-derived ASVs, one was assigned to the chlorophyte family Chlorellaceae and the other to the class Dictyochophyceae (silicoflagellates). Among the 16S rRNA bacterial ASVs, the top 15 contained six assigned to the phylum Bacteroidetes, three each assigned to the phyla Proteobacteria and Actinobacteria and one each assigned to the phyla Marinimicrobia, Verrucomicrobia, and Cyanobacteria. Surprisingly, the cyanobacterial ASV is identical to a Cyanobium sp. strain, Suigetsu-CR5, isolated from a Japanese saline lake (Ohki et al., 2012), while further analysis of other, less abundant cyanobacterial ASVs identified more typical marine Synechococcus strains (see Supplement).
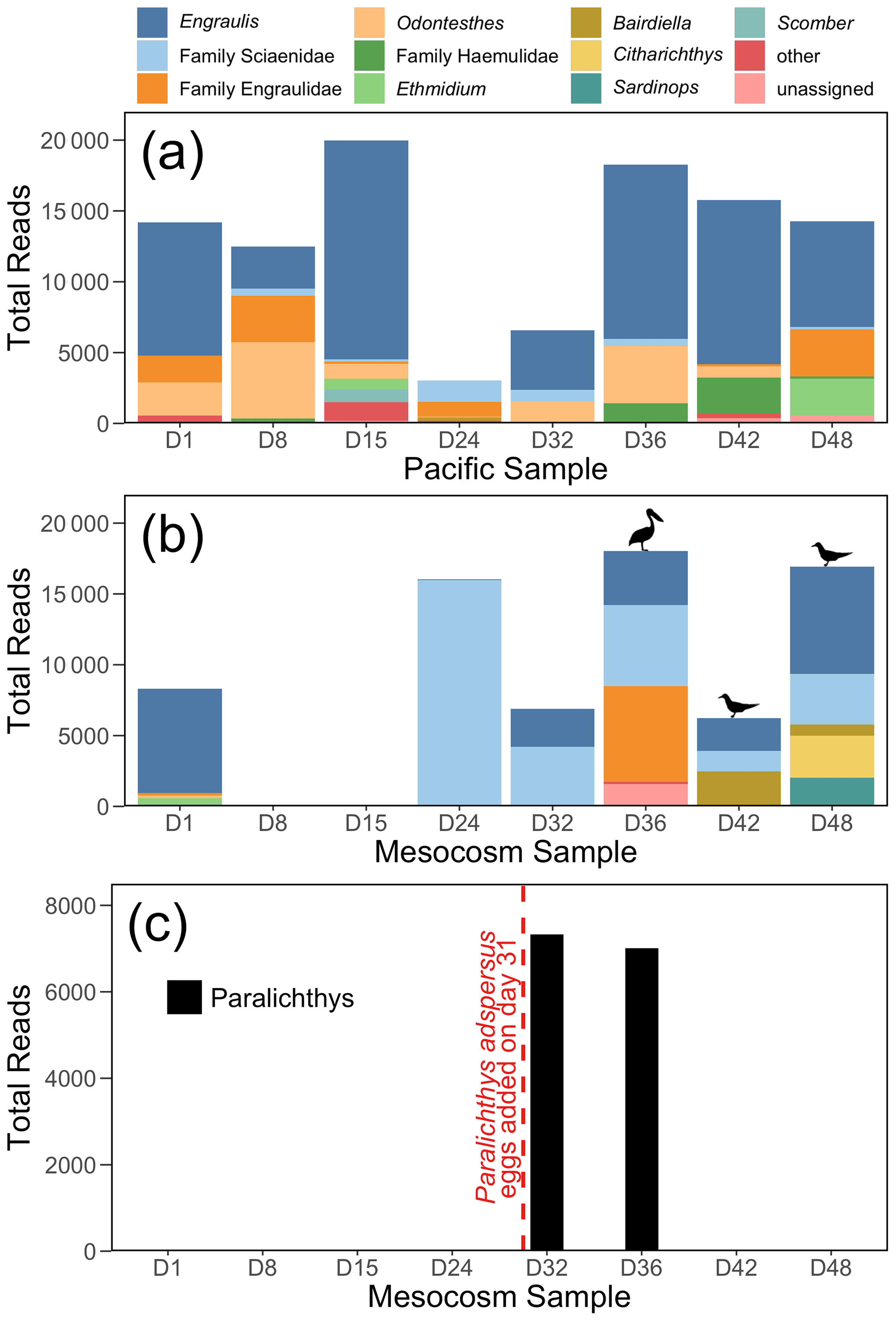
Figure 4The 12S rRNA results, showing top 10 genera (or families, for ASVs that were not annotated to the genus level) for (a) the Pacific and (b) the mesocosm samples, by total number of reads. Detections of Larosterna inca and Pelecanus occidentalis are indicated by the symbols in (b). All reads of Paralichthys have been removed from (a) and (b). Panel (c) shows the total number of reads of Paralichthys, the genus of the fish eggs (Paralichthys adspersus) that were added to the mesocosm on day 31. Because there is no available reference sequence for Paralichthys adspersus, genus-level annotations are used to assess the detections of this species using eDNA.
3.4 Vertebrate detection
The 12S rRNA detected a diversity of vertebrate taxa in the Pacific, dominated by anchovy (genus Engraulis) (Fig. 4a), and with fluctuating numbers of reads. In contrast, the mesocosm showed an initial high number of reads, similarly dominated by anchovy, followed by a drastic drop in reads in the second and third samples (taken on days 8 and 15), which had only 10 and 37 vertebrate reads, respectively (Fig. 4b). The number of vertebrate reads in the mesocosm then increased on day 24 due to reads assigned to the family Sciaenidae (drums and croakers), and subsequently an increase in anchovy reads was also observed. In Fig. 4c, reads assigned to the genus Paralichthys appear on days 32 and 36 following the addition of eggs of fine flounder Paralichthys adspersus on day 31, but are absent in all other samples. Other taxa of note that were detected by 12S rRNA but are not pictured in the above plots because they did not meet the abundance threshold to be included were Inca tern Larosterna inca (six reads each in the mesocosm on days 42 and 48) and brown pelican Pelecanus occidentalis (159 reads in the mesocosm on day 36); the detection of these species is indicated by the corresponding symbols above the bars in Fig. 4b.
The Peruvian eastern boundary upwelling system is known for its dynamic nature (Penven et al., 2005; Huyer et al., 1991; Echevin et al., 2004), reflected by the physicochemical (warming shift, NO fluctuations) and ecological (Fig. 3) changes observed in the unenclosed Pacific ocean over our 50 d sampling period. The variability observed in the Pacific during the sampling period is contrasted by the large shift from these upwelling conditions in the mesocosm, which was isolated from the surrounding Pacific and stratified by NaCl brine injections. This resulted in reduced horizontal and vertical mixing, leading to depletion of nutrients (Fig. 2); NO (Fig. 2c) in particular exhibited a quick and continuous decline, reaching the threshold of detection by day 20. This is in stark contrast to the concentration of NO in the Pacific, which varied from 2.7–19.2 µmol L−1. The observed high concentrations and high day-to-day variability of nitrogen in the natural Pacific samples was indicative of intermittent upwelling, whereby nutrients in the surface waters are replenished from below (Chavez and Messie, 2009; Graco et al., 2017). Si(OH)4 (Fig. 2d) is also lower in the mesocosm and declined in concentration throughout (except for when OMZ water, rich in Si(OH)4, was injected) but never reached levels below 3.4 µmol L−1.
The differences in physical and biogeochemical conditions between the Pacific and the eight mesocosms were pronounced. These physical and chemical differences are reflected in the eDNA metabarcoding comparisons of diversity and the taxonomic composition of the biological communities in the unenclosed Pacific Ocean and one of the mesocosms (M1). The communities responded in a somewhat predictable manner, with those in the Pacific being characteristic of higher-nutrient upwelling conditions and those in the mesocosms being characteristic of lower-nutrient stratified conditions.
4.1 Phytoplankton communities
Phytoplankton are strongly linked to changes in abiotic conditions in upwelling systems, and the connection between environmental conditions and phytoplankton was reflected in the differing communities of the mesocosm and Pacific. Upwelling conditions, as were observed in the Pacific during the experiment, are associated with increased nutrient availability and turbulence (Chavez and Messié, 2009), favoring fast growing non-motile phytoplankton such as diatoms (Margalef, 1978) and newly discovered prasinophyte species (see, e.g., Worden et al., 2004; Simmons et al., 2016). Conversely, relaxing upwelling conditions and subsequent stratification (as were observed in the mesocosm) result in reduced mixing and exhaustion of nutrients (nitrate) in the surface layer. These conditions favor motile organisms like dinoflagellates, who are able to vertically migrate to the deeper nutrient-rich layer at night and back to the surface during the day without the hindrance of turbulence (Margalef, 1978; Smayda, 2010; Smayda and Trainer, 2010).
The stratified mesocosm was dominated by the mixotrophic dinoflagellate Akashiwo sanguinea, a signature bloom species in EBUS (Trainer et al., 2010; Bach et al., 2020). A. sanguinea ASVs were found to be strongly mesocosm-associated in both the COI and 18S rRNA datasets (Fig. 4e and f), and in the 18S rRNA sequences in particular, A. sanguinea was dominant in the mesocosm in the later stages of the experiment, comprising over 50 % of the reads in some samples (Fig. S3). A. sanguinea reads were a minor proportion of the reads in the COI dataset and absent from the 16S rRNA plastid sequences; the disparity in the detection of A. sanguinea by our different PCR primers can be explained by inefficient dinoflagellate amplification by COI primers (Lin et al., 2009) and the loss of most chloroplast genes from dinoflagellates (Koumandou et al., 2004), causing them not to be detected in our 16S rRNA plastidial sequences (Needham and Fuhrman, 2016). While the correlation between cell biomass and amplicon sequence abundance is complicated both by technical biases in metabarcoding and variation in gene copy numbers (Martin et al., 2022), the high biomass of certain taxa as inferred using read numbers is corroborated by other sampling techniques used to sample community composition. In this experiment, A. sanguinea was also identified as the dominant dinoflagellate by imaging flow cytometry and microscopy in mesocosm M1 and in seven of eight mesocosms overall (Bach et al., 2020). Bach et al. (2020) report the common diurnal migration pattern of dinoflagellates, at the surface during the day, migrating to the nutricline at night to take up nutrients. Mixotrophy and vertical migration allow A. sanguinea to exploit nutrients found below the nutrient-depleted surface layer, especially when there is a shallow thermocline (Kudela et al., 2010), as was the case in the mesocosm in this experiment (Fig. 3a in Bach et al., 2020). Blooms of A. sanguinea are regularly observed in EBUS (Du et al., 2011; Kudela et al., 2008; Kahru et al., 2004; Dugdale et al., 1977) under shallow thermocline stratified conditions, following upwelling relaxation. A. sanguinea is typically not found in EBUS phytoplankton communities outside these specific conditions (Kolody et al., 2019; Limardo et al., 2017). Hence, the mesocosm experiment provided the right environmental conditions to initiate a dinoflagellate bloom from the propagules inside the mesocosm, which were either already present in the enclosed water or introduced from the OMZ water additions.
In addition to A sanguinea, many other dinoflagellate ASVs were found to be mesocosm-associated. These dinoflagellate ASVs included the mixotrophic dinoflagellate Fragilidium duplocampanaeforme and its prey, the genus Dinophysis (Park and Kim, 2010). A heterotrophic dinoflagellate known to feed on bloom-forming dinoflagellates, Polykrikos kofoidii (Matsuyama et al., 1999; Tillmann, 2004), was also detected to be mesocosm-associated. Dinoflagellates comprised about two-thirds of the top organisms in our 18S rRNA PC loadings analysis (Fig. 3f), indicating a strong selection of these organisms under stratified conditions and interesting predator–prey relationships. Other mesocosm-associated ASVs included ones annotated to the family Chrysochromulinaceae in the COI and V1–V2 16S rRNA plastidial datasets, which also likely reflect stratification, as blooms of Chrysochromulina have previously been reported in strongly stratified, stable conditions (Edvarsen and Paasche, 1998). Multiple ASVs assigned to the cryptophyte order Pyrenomonadales were found to be mesocosm-associated in the 18S rRNA and V1–V2 16S rRNA plastidial datasets; this could be connected to their green light harvesting phycobiliproteins, which appear to facilitate their growth in low-light conditions (Spear-Bernstein and Miller, 1989), as were found in the mesocosms (Fig. 3c in Bach et al., 2020). However, some Pyrenomondales are predatory mixotrophs; thus, changes in relative abundance could also connect to changes in the bacterial community that provisioned prey resources.
In contrast, the phytoplankton community of the coastal Pacific ocean was dominated by diatoms, typical of communities during higher-nutrient upwelling conditions (Pennington et al., 2006; Chavez et al., 2017; Choi et al., 2020) as well as the cosmopolitan coccolithophore Emiliania huxleyi. As seen by the high levels of NO in the Pacific (Fig. 2c), upwelling conditions were maintained during the period of study, favoring r-selected species like diatoms and maintaining an early succession community (Margalef, 1978; Tyrrell and Merico, 2004). The presence of coccolithophores is indicative of the mixing of some open ocean waters that had come closer to shore due to the coastal El Niño. Although Class II prasinophytes (Mamiellophyceae) were detected in the 16S amplicon data (Fig. S3), they were present at lower relative abundances than the above groups. This is different from observations of prasinophyte importance in more mesotrophic water columns in EBUS off California, USA (Limardo et al., 2017; Kolody et al., 2019; Choi et al., 2020) and off the coast of Chile (De la Iglesia et al., 2020). In the mesocosm, the low levels of nutrients in the mixed layer resulted in lower diatom growth rates, and they sank due to increased stratification (Smayda and Trainer, 2010). Some diatom ASVs remained prevalent in the mesocosm, particularly ASVs assigned to the family Skeletonemataceae (Fig. S3), perhaps as seed populations ready to exploit a change in the environment. In the latter part of the experiment they may also have been seeded by the seabird faeces, given the increased presence of birds in the local area. Diatoms are ubiquitous in anchovy stomachs (Espinoza and Bertrand, 2008), and anchovies are favored by seabirds (Duffy, 1983), and both anchovy and seabird eDNA appeared later in the mesocosm.
There were also three dinoflagellate ASVs common in the Pacific. While one of these was assigned to the family Gymnodiniaceae, a broad family of free-living dinoflagellates, the other two were both assigned to the Syndiniales, a parasitic order (Guillou et al., 2008). Among these two ASVs, one was annotated to the family Duboscquellidae, with some species within this family known to parasitize the tintinnid ciliate Eutintinnus (Coats, 1988). The ciliate Eutintinnus has also been documented to form symbiotic relationships with multiple diatom species (Gómez, 2007, 2020; Vincent et al., 2018). Interestingly, an ASV assigned to the genus Eutintinnus was also found in the Pacific, suggesting that both the parasite–host relationship and the symbiotic relationship of this ciliate was found in the ocean surrounding the mesocosms.
An unusual and abundant ASV identical to Cyanobium sp. Suigetsu-CR5 (Ohki et al., 2012) was found in the coastal Pacific samples. The primers used to amplify 16S rRNA did not provide full-length 16S rRNA gene sequences to confirm that this ASV was the same organism as that isolated from a Japanese lake. However, Cyanobium sp. Suigetsu-CR5-like sequences have been reported from the open ocean northeastern Pacific (Sudek et al., 2015). Taken together, these results suggest that a marine Cyanobium sp. Suigetsu-CR5-like organism exists and given the right environmental conditions can become a dominant cyanobacterium.
4.2 Zooplankton communities
The mesozooplankton communities of the mesocosm and Pacific as detected via COI were differentiated primarily by Pacific-associated ASVs assigned to the rotifer family Synchaetidae and the calanoid copepod family Acartiidae (Fig. 3e); these results are in agreement with vertical tows of an Apstein net of mesh size 100 µm during the experiment (unpublished data). Rotifers were detected initially in similar abundance in both the mesocosm and the Pacific in the COI data, but declined in abundance in the mesocosm while remaining more abundant, albeit highly variable, in the Pacific (Fig. S3b). Conversely, ASVs assigned to the copepod family Acartiidae were more abundant in the Pacific than the mesocosm from the start of the experiment in both the net tow and COI data (unpublished data, Fig. S3b), indicating that fewer individuals of this family were trapped within the mesocosm when it was closed, owing to the patchy distribution of zooplankton (Wiebe and Holland, 1968). The most dominant zooplankton taxa found were the calanoid copepod Paracalanus (detected by COI and 18S) and the cyclopoid copepod Hemicyclops (detected only by 18S); however, these taxa were found in similar abundances in both the mesocosm and Pacific samples throughout the experiment by both eDNA metabarcoding and net tow data and thus did not differentiate the two sampling sites (Fig. S3b and c). Copepods have lifespans that are greater than the duration of the experiment (Ianora, 1998), so a significant response of the copepod communities was not captured within the mesocosm relative to the Pacific. Longer-term experiments will be required to study responses of animals with generation times of months or greater.
The nanozooplankton community of the mesocosm was separated from that of the Pacific by the higher relative abundance of the family Cafeteriaceae, the higher relative abundance of the cercozoan genus Protaspis, and the relative lower abundance of the families Paulinellidae and Vexilliferidae. The family Cafeteriaceae is a made up of heterotrophic nanoflagellates, many of which are filter-feeding bacterivores (Schoenle et al., 2020). Their increased abundance in the mesocosm may reflect their tendency to associate with detritus (Patterson et al., 1993) or adhere to structure (Baker et al., 2018; Boenigk and Arndt, 2000), as was available in the mesocosm. The amoebozoan family Vexilliferidae is ubiquitous in both marine and estuarine environments (Page, 1983), while the amoeboid family Paulinellidae (phylum Cercozoa) contains phototrophic and heterotrophic species which inhabit freshwater, brackish, and marine environments (Kim and Park, 2016).
4.3 Heterotrophic and photosynthetic bacterial community
As seen in the beta diversity analyses in Fig. 3, the bacterial community of the mesocosm diverged from that of the Pacific faster than the communities detected by COI, 18S rRNA, and 16S plastidial sequences, separating on PC1 by day 8 rather than day 15. This may be at least partially explained by the fast growth rate of prokaryotic cells (Zubkov, 2014). The ASVs that were Pacific-associated were representative of typical coastal, phytoplankton-rich communities (Needham and Fuhrman, 2016; Buchan et al., 2014). These ASVs included an ASV assigned to the 16S rRNA clade SAR86, one of the most abundant constituents of microbial communities in the surface ocean (Dupont et al., 2012), and an ASV assigned to the family Synechococcaceae, a ubiquitous family of cyanobacteria that is most abundant in nutrient-rich surface waters (Partensky et al., 1999), as was found in the Pacific during the course of the experiment (Fig. 2). Mesocosm-associated ASVs included the methylotrophic family Methylophagaceae (Neufeld et al., 2007), Pseudohongiella, a genus recently isolated from the northwest Pacific (Xu et al., 2016; Park et al., 2014), multiple ASVs of the family Rhodobacteraceae, and an ASV assigned to the NS5 marine group. Rhodobacteraceae have previously been documented to increase in response to a bloom of A. sanguinea (Yang et al., 2012), as was documented within the mesocosm, and have also been documented to increase in blooms of the harmful dinoflagellate Alexandrium (Hattenrath-Lehmann and Gobler, 2017). Similarly, the NS5 marine group has previously been documented to have increased in abundance with blooms of both A. sanguinea (Yang et al., 2015) and Alexandrium (Hattenrath-Lehmann and Gobler, 2017).
One of the most unusual taxa that was detected was from the order Saccharimonadales within the superphylum Cand. Patescibacteria (Parks et al., 2018), which was first detected in M1 on day 15 and in the Pacific on day 24 and subsequently increased in abundance in both M1 and the Pacific. The rise corresponded with an increase in small bacteria and heterotrophic bacteria overall in flow cytometry samples. While this group (Saccharibacteria) has been detected in seawater previously (Hugenholtz et al., 2001) and is commonly found across many different environments from soil to the human gut and oral microbiomes (Kuehbacher et al., 2008; Marcy et al., 2007; Ferrari et al., 2005), some representatives of this group have an unusual symbiotic lifestyle. They were recently isolated from the human oral microbiome where they were found to be epibionts of Actinobacteria (He et al., 2015). Recent genomic evidence reveals that Saccharibacteria have small genomes (<1 Mb), with genomic contents consistent with a symbiotic lifestyle (Lemos et al., 2019), and microscopy and filter size fractionation have found them to be very small cells (or “ultra-small bacteria”). To account for their rapid rise in prevalence, it is possible that these Saccharibacteria colonized the mesocosms via a biofilm (outside and inside the experiment itself) or were introduced via seabird faeces, as Saccharibacteria have previously been detected in the avian microbiome (Hird et al., 2015). The timing of the increase is consistent with the appearance of the seabirds on the mesocosms. Since the seabirds were foraging in the vicinity of the mesocosms, it is possible that they may have contributed to the increase in the Pacific samples as well. In any case, this observation warrants further study to understand the lifestyle and genomic potential of this enigmatic group.
4.4 Insights into vertebrates
The 12S rRNA metabarcoding data detected distinctly different communities in the Pacific and in the mesocosm. Bony fish typical of the coastal Peruvian upwelling system, dominated by the Peruvian anchoveta (Engraulis ringens), were found in the Pacific samples. In the mesocosm, the species assemblage reflected three experimental perturbations: (1) the exclusion of vertebrates via a 3 mm mesh, (2) the addition of eggs of Paralichthys adspersus, and (3) the impact of resting seabirds. During the deployment of the mesocosm, the initial water was filtered through a net of mesh size 3 mm, and collection of the OMZ water that was later added was conducted through a net of mesh size 10 mm. The fine mesh sizes effectively excluded most, if not all, living vertebrate stages from the enclosure but not their eDNA. The initial vertebrate eDNA in the mesocosm decayed rapidly, as evidenced by the extremely low read counts on days 8 and 15 (10 and 37 vertebrate reads, respectively). The virtual disappearance of vertebrate eDNA a week after the start of the experiment is in line with estimates of bony fish eDNA decay rates (Sassoubre et al., 2016), which show an exponential decline in eDNA concentrations with time. Quantification of eDNA decay rates of northern anchovy (Engraulis mordax), which is congeneric with the Peruvian anchoveta E. ringens, showed that E. mordax DNA concentration reached the threshold of detection by a qPCR assay within three days of the fish being removed from the enclosure (Sassoubre et al., 2016). There may have been some eDNA introduced by the addition of the OMZ water; however, the 10-day period between when this water was collected on day 5 and when the first eDNA sampling occurred following OMZ water addition (day 15) coupled with the decay rate of eDNA makes the detection of this eDNA unlikely.
The addition of eggs of the fish Paralichthys adspersus (fine flounder) to the mesocosm on day 31 led to a notable spike in Paralichthys reads on days 32 and 36; Paralichthys was not detected in any other samples in the mesocosm or Pacific. Because P. adspersus does not have a reference sequence available in GenBank (release 238.0) for the region targeted by the MiFish primers, no species-level detections were possible, and thus we assessed the detection of this species by eDNA through annotations to the genus Paralichthys. The strong signal of Paralichthys in the 12S rRNA results is particularly notable because the introduction of fish eggs was not detected via net sampling (Bach et al., 2020). The fish eggs apparently did not develop and decomposed or sank out of the mesocosm fairly quickly. Based on their size, the estimated sinking rate for P. adspersus eggs, which average a diameter of 0.66–0.80 mm (Silva and Oliva, 2010), is about 5 mm s−1 (Robertson, 1981). Additionally, for any eggs that hatched, larval settling rates for two other species of Paralichthys is estimated to be about 10 mm s−1 (Hare et al., 2006). With the very stable, stratified waters of the mesocosm, the lack of turbulence likely would have caused any eggs or larvae to quickly settle out of the water column, and coupled with eDNA decay rates, it is not surprising that this signal was no longer detected in the eDNA sample taken on day 42.
The final and most pronounced influence on the vertebrate composition within the mesocosm was the impact of seabirds, which occasionally rested on the mesocosms until day 36 but increased abruptly in abundance thereafter (Bach et al., 2020). DNA from both seabirds and their prey was detected in the second half of the experiment. Inca tern (Larosterna inca), the most common bird observed on the mesocosms, was detected in the last two mesocosm samples (days 42 and 48), while brown pelican (Pelecanus occidentalis) was detected in the mesocosm on day 36 (Fig. 4b). While bony fish are the intended target of the MiFish primers, these primers are also capable of detecting other vertebrates due to sequence similarity in the targeted region of 12S rRNA gene, albeit with weaker amplification (Miya et al., 2015; Monuki et al., 2021). As such, while we detected seabirds using our primers, they are likely not well represented in our data; primers that specifically target birds should improve assessments of their eDNA (Ushio et al., 2018). Inca terns are known to feed primarily on E. ringens, and the defecation by these seabirds into the mesocosm is inferred by the increase in Engraulis DNA in the last four samples. Additionally, many reads assigned to the family Sciaenidae (drums and croakers) were detected in the mesocosm, but because of the lack of reference sequences for most of the species in this family that are found in the coastal waters of Peru, it is impossible to determine the identity of these species. However, members of the family Sciaenidae have been documented to be important prey species for seabirds in other regions (Lamb et al., 2017). In addition, the increase in small bacteria (Fig. 2j) in both the mesocosm and the Pacific may be a result of the seabirds defecating in and around the mesocosm.
4.5 Strengths and limitations of eDNA metabarcoding in mesocosm experiments
Through the use of eDNA metabarcoding targeting multiple genetic loci, we were able to examine community-level changes, detecting 12 244 ASVs assigned to 85 unique phyla; amongst these ASVs, the taxonomy of 816 ASVs could be resolved to the genus level. Sampling methods commonly used to assess communities often inherently size-select (vertical net tows; Skjoldal et al., 2013), are hindered by both the labor-intensive nature of morphological assessments and the inherent variation in expertise amongst taxonomists (Harvey et al., 2017), and are limited in the taxonomic resolution they provide (e.g., flow cytometry). eDNA metabarcoding provides a method that overcomes some of these limitations: by sampling genetic material in seawater, there is higher sensitivity for detection, there is no minimum size threshold (as there is in light microscopy or net tows), and metabarcoding of genetic material provides objective taxonomic assignments at higher resolution than morphological identification (Berry et al., 2015), depending on the variable region sequenced and its efficacy for resolving different taxonomic groups (Wear et al., 2018; Parada et al., 2016).
eDNA metabarcoding also comes with a number of challenges and limitations, particularly the issue of incomplete reference databases. For example, amongst the 88 COI ASVs that met the criteria for inclusion in the analysis of PC1 loading scores (Fig. 3e–h), only 17 had a match greater than 95 % to a reference sequence. Most of the COI ASVs identified as driving differences between the mesocosm and Pacific samples and visualized in Fig. 3e had percent matches to reference sequences of 80 %–90 % and thus were either annotated to higher orders of taxonomy or left unassigned; the only exceptions to this are the ASVs assigned to Akashiwo sanguinea, Emiliania huxleyi, Skeletonema, and Class V prasinophytes. A similar issue occurred with the 12S rRNA data – because of the lack of reference sequences for Peruvian fish, most ASVs could only be annotated to the genus or family level. While reference databases are constantly improving thanks to large-scale sequencing efforts (e.g., Rimet et al., 2016; Gold et al., 2021), the lack of reference sequences will continue to hinder metabarcoding efforts, especially for uncharismatic and cryptic taxa.
4.6 Summary and conclusion
In this study multiple marker eDNA metabarcodes were used to follow the evolution of diverse communities in an in situ mesocosm and the nearby unenclosed Pacific Ocean over 50 d. A quick divergence between mesocosm and open ocean communities was observed at multiple trophic levels. The mesocosm community evolved quickly into one that is common in stable, stratified conditions (Smayda and Trainer, 2010; Margalef, 1978). Spilling et al. (2018) have noted that in the Baltic, dinoflagellates respond to increases in stratification driven by either changes in salinity or temperature. The Pacific community retained the character of communities found under nutrient-replete upwelling conditions. Upwelling conditions are associated with increased nutrient availability and turbulence (Chavez and Messié, 2009), favoring fast-growing, non-motile phytoplankton such as diatoms (Margalef, 1978). Conversely, weak upwelling and increased stratification result in reduced mixing and removal of nutrients from the shallow mixed layer (Smayda and Trainer, 2010), favoring motile organisms like dinoflagellates who are able to migrate to the nutricline at night without the hindrance of turbulence (Smayda, 2010; Margalef, 1978). The bacterial communities reflected the changes in phytoplankton composition and likely the influence of seabirds. Many of the abundant bacterial taxa in the mesocosm are reported to increase in response to blooms of dinoflagellates, while typical open-ocean cyanobacteria (Synechococcaceae) and heterotrophic bacteria (e.g., SAR86) were more abundant in the Pacific. The increase in resting seabirds towards the end of the experiment led to unusual bacterial groups that have been reported to be present in the guts of animals (namely the Saccharibacteria). Primary and secondary consumers in upwelling ecosystems, such as zooplankton and fish, have slower responses to physicochemical conditions, so their responses were incompletely resolved in the 50 d experiment.
The method of eDNA metabarcoding is rapidly evolving, facing a series of challenges and opportunities. For example, incomplete reference database issues hindered the confidence of our taxonomic assignments. This was particularly notable off the coast of Peru, where few species have been well-characterized genetically. Nonetheless, the multiple-marker eDNA metabarcoding results presented here show how single samples can yield results that are comparable and complementary to a multitude of traditional and other emerging methods to survey marine biodiversity across the tree of life. The application of multiple genetic markers provided insight into how multiple trophic levels interact under changing physical and biological (seabirds) conditions, revealing coupled changes in bacterial (16S), phytoplankton (18S), zooplankton (COI), and vertebrate (12S) communities. They also revealed evidence of potential predator–prey and parasite–host relationships, whose complexity could be explored further in interaction networks.
The effects of perturbations, either purposeful (additions of OMZ water, brine solution, and fish eggs) or unintended (seabird droppings), on marine communities were clearly resolved with our methods. The perturbations provided new insights into ecosystem processes that are difficult to study otherwise. Mesocosm experiments are challenging because of the difficulties of reproducing physical conditions in contained systems. However, fundamental ecosystem processes in the ocean (i.e., stratification) can be well studied with mesocosms as described here. The selection of dinoflagellates under the combined effect of the salinity-driven stratified conditions in the mesocosm and the warm conditions brought about by the coastal El Niño (which affected both the mesocosm and Pacific) may be an indication of how EBUS could respond to the global environmental changes (in both salinity and temperature) forecast for the future (Pachauri et al., 2014). In support of this, evidence from the fossil record indicates that during the Paleocene thermal maximum, when temperatures where warmer than present, there was a global response of surface-dwelling coastal dinoflagellate communities (Crouch et al., 2001). Mesocosm experiments, like the one studied here, provide a valuable complement to traditional observational studies of upwelling systems. Insights from the genetic methods applied here will guide us towards a more mechanistic understanding of the processes that are shaping EBUS communities in a changing ocean.
All code and data required to reproduce the analytical results, figures, and tables for this study are available on GitHub at https://github.com/MBARI-BOG/KOSMOS_eDNA_paper (last access: 21 February 2023, https://doi.org/10.5281/Zenodo.7255826, Min and Pitz, 2022).
The supplement related to this article is available online at: https://doi.org/10.5194/bg-20-1277-2023-supplement.
MAM, DMN, SS, NKT, KJP, AZW, and FPC wrote the paper. MAM and DMN conducted the data analysis. GMC led the collection and preservation of eDNA samples with assistance from DMN and CP. CP led the collection and preservation of flow cytometry samples with assistance from GMC and DMN. BG analyzed the flow cytometry data. EvdE analyzed the nutrients. UR and AL designed the overall mesocosm experiment. FPC and AZW designed the sampling/analyses of samples described in the paper and secured funding to carry out the work.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Ecological and biogeochemical functioning of the coastal upwelling system off Peru: an in situ mesocosm study”. It is not associated with a conference.
We thank the members of the GEOMAR team and Carlos Robles from IMARPE for providing logistical, physical, and moral support during the experiment.
This research has been supported by the Gordon and Betty Moore Foundation (grant no. GBMF 3788); the National Aeronautics and Space Administration, Earth Sciences Division (grant no. NNX14AP62A); and the National Science Foundation, Directorate for Biological Sciences (grant no. NSF DEB-1639033).
This paper was edited by Hans-Peter Grossart and reviewed by two anonymous referees.
Agarwala, R., Barrett, T., Beck, J., Benson, D. A., Bollin, C., Bolton, E., Bourexis, D., Brister, J. R., Bryant, S. H., Canese, K., Cavanaugh, M., Charowhas, C., Clark, K., Dondoshansky, I., Feolo, M., Fitzpatrick, L., Funk, K., Geer, L. Y., Gorelenkov, V., Graeff, A., Hlavina, W., Holmes, B., Johnson, M., Kattman, B., Khotomlianski, V., Kimchi, A., Kimelman, M., Kimura, M., Kitts, P., Klimke, W., Kotliarov, A., Krasnov, S., Kuznetsov, A., Landrum, M. J., Landsman, D., Lathrop, S., Lee, J. M., Leubsdorf, C., Lu, Z., Madden, T. L., Marchler-Bauer, A., Malheiro, A., Meric, P., Karsch-Mizrachi, I., Mnev, A., Murphy, T., Orris, R., Ostell, J., O'Sullivan, C., Palanigobu, V., Panchenko, A. R., Phan, L., Pierov, B., Pruitt, K. D., Rodarmer, K., Sayers, E. W., Schneider, V., Schoch, C. L., Schuler, G. D., Sherry, S. T., Siyan, K., Soboleva, A., Soussov, V., Starchenko, G., Tatusova, T. A., Thibaud-Nissen, F., Todorov, K., Trawick, B. W., Vakatov, D., Ward, M., Yaschenko, E., Zasypkin, A., and Zbicz, K.: Database resources of the National Center for Biotechnology Information, Nucl. Acids Res., 46, D8–D13, https://doi.org/10.1093/nar/gkx1095, 2018.
Amaral-Zettler, L. A., McCliment, E. A., Ducklow, H. W., and Huse, S. M.: A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA Genes, PLoS One, 4, 1–9, https://doi.org/10.1371/journal.pone.0006372, 2009.
Amaral-Zettler, L. A., Bauer, M., Berg-Lyons, D., Betley, J., Caporaso, J. G., Ducklow, H. W., Fierer, N., Fraser, L., Gilbert, J. A., Gormley, N., Huntley, J., Huse, S. M., Jansson, J. K., Jarman, S. N., Knight, R., Lau, C. L., and Walters, W. A.: EMP 18S Illumina Amplicon Protocol, protocols.io, https://doi.org/10.17504/protocols.io.nuvdew6, 2018.
Ayón, P., Swartzman, G., Bertrand, A., Gutiérrez, M., and Bertrand, S.: Zooplankton and forage fish species off Peru: Large-scale bottom-up forcing and local-scale depletion, Prog. Oceanogr., 79, 208–214, https://doi.org/10.1016/j.pocean.2008.10.023, 2008.
Bach, L. T., Paul, A. J., Boxhammer, T., von der Esch, E., Graco, M., Schulz, K. G., Achterberg, E., Aguayo, P., Arístegui, J., Ayón, P., Baños, I., Bernales, A., Boegeholz, A. S., Chavez, F., Chavez, G., Chen, S. M., Doering, K., Filella, A., Fischer, M., Grasse, P., Haunost, M., Hennke, J., Hernández-Hernández, N., Hopwood, M., Igarza, M., Kalter, V., Kittu, L., Kohnert, P., Ledesma, J., Lieberum, C., Lischka, S., Löscher, C., Ludwig, A., Mendoza, U., Meyer, J., Meyer, J., Minutolo, F., Cortes, J. O., Piiparinen, J., Sforna, C., Spilling, K., Sanchez, S., Spisla, C., Sswat, M., Moreira, M. Z., and Riebesell, U.: Factors controlling plankton community production, export flux, and particulate matter stoichiometry in the coastal upwelling system off Peru, Biogeosciences, 17, 4831–4852, https://doi.org/10.5194/bg-17-4831-2020, 2020.
Bachy, C., Wittmers, F., Muschiol, J., Hamilton, M., Henrissat, B., and Worden, A. Z.: The Land-Sea Connection: Insights Into the Plant Lineage from a Green Algal Perspective, Annu. Rev. Plant Biol., 73, 585–616, https://doi.org/10.1146/annurev-arplant-071921-100530, 2022.
Baker, P., Minzlaff, U., Schoenle, A., Schwabe, E., Hohlfeld, M., Jeuck, A., Brenke, N., Prausse, D., Rothenbeck, M., Brix, S., Frutos, I., Jörger, K. M., Neusser, T. P., Koppelmann, R., Devey, C., Brandt, A., and Arndt, H.: Potential contribution of surface-dwelling Sargassum algae to deep-sea ecosystems in the southern North Atlantic, Deep-Sea Res. Pt. II, 148, 21–34, https://doi.org/10.1016/j.dsr2.2017.10.002, 2018.
Berry, O., Bulman, C., Bunce, M., Coghlan, M., Murray, D. C., and Ward, R. D.: Comparison of morphological and DNA metabarcoding analyses of diets in exploited marine fishes, Mar. Ecol. Prog. Ser., 540, 167–181, https://doi.org/10.3354/meps11524, 2015.
Boenigk, J. and Arndt, H.: Particle handling during interception feeding by four species of heterotrophic nanoflagellates, J. Eukaryot. Microbiol., 47, 350–358, https://doi.org/10.1111/j.1550-7408.2000.tb00060.x, 2000.
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., Alexander, H., Alm, E. J., Arumugam, M., Asnicar, F., Bai, Y., Bisanz, J. E., Bittinger, K., Brejnrod, A., Brislawn, C. J., Brown, C. T., Callahan, B. J., Caraballo-Rodríguez, A. M., Chase, J., Cope, E. K., Da Silva, R., Diener, C., Dorrestein, P. C., Douglas, G. M., Durall, D. M., Duvallet, C., Edwardson, C. F., Ernst, M., Estaki, M., Fouquier, J., Gauglitz, J. M., Gibbons, S. M., Gibson, D. L., Gonzalez, A., Gorlick, K., Guo, J., Hillmann, B., Holmes, S., Holste, H., Huttenhower, C., Huttley, G. A., Janssen, S., Jarmusch, A. K., Jiang, L., Kaehler, B. D., Kang, K. Bin, Keefe, C. R., Keim, P., Kelley, S. T., Knights, D., Koester, I., Kosciolek, T., Kreps, J., Langille, M. G. I., Lee, J., Ley, R., Liu, Y. X., Loftfield, E., Lozupone, C., Maher, M., Marotz, C., Martin, B. D., McDonald, D., McIver, L. J., Melnik, A. V., Metcalf, J. L., Morgan, S. C., Morton, J. T., Naimey, A. T., Navas-Molina, J. A., Nothias, L. F., Orchanian, S. B., Pearson, T., Peoples, S. L., Petras, D., Preuss, M. L., Pruesse, E., Rasmussen, L. B., Rivers, A., Robeson, M. S., Rosenthal, P., Segata, N., Shaffer, M., Shiffer, A., Sinha, R., Song, S. J., Spear, J. R., Swafford, A. D., Thompson, L. R., Torres, P. J., Trinh, P., Tripathi, A., Turnbaugh, P. J., Ul-Hasan, S., van der Hooft, J. J. J., Vargas, F., Vázquez-Baeza, Y., Vogtmann, E., von Hippel, M., Walters, W., Wan, Y., Wang, M., Warren, J., Weber, K. C., Williamson, C. H. D., Willis, A. D., Xu, Z. Z., Zaneveld, J. R., Zhang, Y., Zhu, Q., Knight, R., and Caporaso, J. G.: Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2, Nat. Biotechnol., 37, 852–857, https://doi.org/10.1038/s41587-019-0209-9, 2019.
Boussarie, G., Bakker, J., Wangensteen, O. S., Mariani, S., Bonnin, L., Juhel, J. B., Kiszka, J. J., Kulbicki, M., Manel, S., Robbins, W. D., Vigliola, L., and Mouillot, D.: Environmental DNA illuminates the dark diversity of sharks, Sci. Adv., 4, 5, https://doi.org/10.1126/sciadv.aap9661, 2018.
Buchan, A., LeCleir, G. R., Gulvik, C. A., and González, J. M.: Master recyclers: features and functions of bacteria associated with phytoplankton blooms, Nat. Rev. Microbiol., 12, 686–698, https://doi.org/10.1038/nrmicro3326, 2014.
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P.: DADA2: High-resolution sample inference from Illumina amplicon data, Nat. Methods, 13, 581–583, https://doi.org/10.1038/nmeth.3869, 2016.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, T. L.: BLAST+: Architecture and applications, BMC Bioinformatics, 10, 1–9, https://doi.org/10.1186/1471-2105-10-421, 2009.
Carr, M. E.: Estimation of potential productivity in Eastern Boundary Currents using remote sensing, Deep-Sea Res. Pt. II, 49, 59–80, https://doi.org/10.1016/S0967-0645(01)00094-7, 2001.
Carr, M. E. and Kearns, E. J.: Production regimes in four Eastern Boundary Current systems, Deep-Sea Res. Pt. II, 50, 3199–3221, https://doi.org/10.1016/j.dsr2.2003.07.015, 2003.
Chavez, F. P. and Messié, M.: A comparison of Eastern Boundary Upwelling Ecosystems, Prog. Oceanogr., 83, 80–96, https://doi.org/10.1016/j.pocean.2009.07.032, 2009.
Chavez, F. P., Pennington, J. T., Michisaki, R. P., Blum, M., Chavez, G. M., Friederich, J., Jones, B., Herlien, R., Kieft, B., and Hobson, B.: Climate variability and change: response of a coastal ocean ecosystem, Oceanography, 30, 128–145, 2017.
Choi, C. J., Jimenez, V., Needham, D. M., Poirier, C., Bachy, C., Alexander, H., Wilken, S., Chavez, F. P., Sudek, S., Giovannoni, S. J., and Worden, A. Z.: Seasonal and geographical transitions in eukaryotic phytoplankton community structure in the Atlantic and Pacific Oceans, Front. Microbiol., 11, 542372, https://doi.org/10.3389/fmicb.2020.542372, 2020.
Closek, C., Djurhuus, A., Pitz, K., Kelly, R., Michisaki, R., Walz, K., Starks, H., Chavez, F., Boehm, A., and Breitbart, M.: Environmental DNA (eDNA) 18S metabarcoding Illumina MiSeq NGS PCR Protocol, protocols.io, https://doi.org/10.17504/protocols.io.n2vdge6, 2018a.
Closek, C., Djurhuus, A., Pitz, K., Kelly, R., Michisaki, R., Walz, K., Starks, H., Chavez, F., Boehm, A., and Breitbart, M.: Environmental DNA (eDNA) COI metabarcoding Illumina MiSeq NGS PCR Protocol, protocols.io, https://doi.org/10.17504/protocols.io.mwnc7de, 2018b.
Coats, D. W.: Duboscquella cachoni N. Sp., a Parasitic Dinoflagellate Lethal to Its Tintinnine Host Eutintinnus pectinis 1, J. Protozool., 35, 607–617, 1988.
Crouch, E. M., Heilmann-Clausen, C., Brinkhuis, H., Morgans, H. E. G., Rogers, K. M., Egger, H., and Schmitz, B.: Global dinoflagellate event associated with the late Paleocene thermal maximum, Geology, 29, 315–318, https://doi.org/10.1130/0091-7613(2001)029<0315:GDEAWT>2.0.CO;2, 2001.
Daims, H., Brühl, A., Amann, R., Schleifer, K. H., and Wagner, M.: The domain-specific probe EUB338 is insufficient for the detection of all bacteria: Development and evaluation of a more comprehensive probe set, Syst. Appl. Microbiol., 22, 434–444, https://doi.org/10.1016/S0723-2020(99)80053-8, 1999.
Decelle, J., Romac, S., Stern, R. F., Bendif, E. M., Zingone, A., Audic, S., Guiry, M. D., Guillou, L., Tessier, D., Le Gall, F., Gourvil, P., Dos Santos, A. L., Probert, I., Vaulot, D., de Vargas, C., and Christen, R.: PhytoREF: A reference database of the plastidial 16S rRNA gene of photosynthetic eukaryotes with curated taxonomy, Mol. Ecol. Resour., 15, 1435–1445, https://doi.org/10.1111/1755-0998.12401, 2015.
De La Iglesia, R., Echenique-Subiabre, I., Rodríguez-Marconi, S., Espinoza, J. P., Von Dassow, P., Ulloa, O., and Trefault, N.: Distinct oxygen environments shape picoeukaryote assemblages thriving oxygen minimum zone waters off central Chile, J. Plankton Res., 42, 514–529, https://doi.org/10.1093/plankt/fbaa036, 2020.
Didion, J. P., Martin, M., and Collins, F. S.: Atropos: Specific, sensitive, and speedy trimming of sequencing reads, PeerJ, 2017, 1–19, https://doi.org/10.7717/peerj.3720, 2017.
Djurhuus, A., Pitz, K., Sawaya, N. A., Rojas-Márquez, J., Michaud, B., Montes, E., Muller-Karger, F., and Breitbart, M.: Evaluation of marine zooplankton community structure through environmental DNA metabarcoding, Limnol. Oceanogr. Method., 16, 209–221, https://doi.org/10.1002/lom3.10237, 2018.
Du, X., Peterson, W., McCulloch, A., and Liu, G.: An unusual bloom of the dinoflagellate Akashiwo sanguinea off the central Oregon, USA, coast in autumn 2009, Harmful Algae, 10, 784–793, https://doi.org/10.1016/j.hal.2011.06.011, 2011.
Duffy, D. C.: The foraging ecology of Peruvian seabirds, Auk, 100, 800–810, 1983.
Dugdale, R. C., Goering, J. J., Barber, R. T., Smith, R. L., and Packard, T. T.: Denitrification and hydrogen sulfide in the Peru upwelling region during 1976, Deep-Sea Res., 24, 601–608, 1977.
Dupont, C. L., Rusch, D. B., Yooseph, S., Lombardo, M. J., Alexander Richter, R., Valas, R., Novotny, M., Yee-Greenbaum, J., Selengut, J. D., Haft, D. H., Halpern, A. L., Lasken, R. S., Nealson, K., Friedman, R., and Craig Venter, J.: Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage, ISME J., 6, 1186–1199, https://doi.org/10.1038/ismej.2011.189, 2012.
Echevin, V., Puillat, I., Grados, C., and Dewitte, B.: Seasonal and mesoscale variability in the Peru Upwelling System from in situ data during the years 2000 to 2004, Gayana (Concepción), 68, 167–173, https://doi.org/10.4067/s0717-65382004000200031, 2004.
Edvarsen, B. and Paasche, E.: Bloom dynamics and physiology of Prymnesium and Chrysochromulina, Physiol. Ecol. Harmful Algal Bloom., 41, 193–208, 1998.
Espinoza, P. and Bertrand, A.: Revisiting Peruvian anchovy (Engraulis ringens) trophodynamics provides a new vision of the Humboldt Current system, Prog. Oceanogr., 79, 215–227, https://doi.org/10.1016/j.pocean.2008.10.022, 2008.
Evans, N. T., Olds, B. P., Renshaw, M. A., Turner, C. R., Li, Y., Jerde, C. L., Mahon, A. R., Pfrender, M. E., Lamberti, G. A., and Lodge, D. M.: Quantification of mesocosm fish and amphibian species diversity via environmental DNA metabarcoding, Mol. Ecol. Resour., 16, 29–41, https://doi.org/10.1111/1755-0998.12433, 2016.
Ferrari, B. C., Binnerup, S. J., and Gillings, M.: Microcolony cultivation on a soil substrate membrane system selects for previously uncultured soil bacteria, Appl. Environ. Microbiol., 71, 8714–8720, https://doi.org/10.1128/AEM.71.12.8714-8720.2005, 2005.
Folmer, O., Hoeh, W. R., Black, M. B., and Vrijenhoek, R. C.: Conserved primers for PCR amplification of mitochondrial DNA from different invertebrate phyla, Mol. Mar. Biol. Biotechnol., 3, 294–299, 1994.
Garreaud, R. D.: A plausible atmospheric trigger for the 2017 coastal El Niño, Int. J. Climatol., 38, e1296–e1302, https://doi.org/10.1002/joc.5426, 2018.
Giovannoni, S. J., Britschgi, T. B., Moyer, C. L., and Field, K. G.: Genetic diversity in Sargasso Sea bacterioplankton, Nature, 345, 60–63, https://doi.org/10.1038/345060a0, 1990.
Gold, Z., Curd, E., Goodwin, K., Choi, E., Frable, B., Thompson, A., Burton, R., Kacev, D., and Barber, P.: Improving metabarcoding taxonomic assignment: A case study of fishes in a large marine ecosystem, Mol. Ecol. Resour., 21, 2546–2564, https://doi.org/10.22541/au.161407483.33882798/v1, 2021.
Gómez, F.: On the consortium of the tintinnid Eutintinnus and the diatom Chaetoceros in the Pacific Ocean, Mar. Biol., 151, 1899–1906, https://doi.org/10.1007/s00227-007-0625-0, 2007.
Gómez, F.: Symbiotic interactions between ciliates (Ciliophora) and diatoms (Bacillariophyceae), Rev. Biol. Trop., 68, 2020.
Graco, M. I., Purca, S., Dewitte, B., Castro, C. G., Morón, O., Ledesma, J., Flores, G., and Gutiérrez, D.: The OMZ and nutrient features as a signature of interannual and low-frequency variability in the Peruvian upwelling system, Biogeosciences, 14, 4601–4617, https://doi.org/10.5194/bg-14-4601-2017, 2017.
Gruber, N.: Warming up, turning sour, losing breath: Ocean biogeochemistry under global change, Philos. T. R. Soc. A, 369, 1980–1996, https://doi.org/10.1098/rsta.2011.0003, 2011.
Guillou, L., Viprey, M., Chambouvet, A., Welsh, R. M., Kirkham, A. R., Massana, R., Scanlan, D. J., and Worden, A. Z.: Widespread occurrence and genetic diversity of marine parasitoids belonging to Syndiniales (Alveolata), Environ. Microbiol., 10, 3349–3365, https://doi.org/10.1111/j.1462-2920.2008.01731.x, 2008.
Hare, J. A., Walsh, H. J., and Wuenschel, M. J.: Sinking rates of late-stage fish larvae: Implications for larval ingress into estuarine nursery habitats, J. Exp. Mar. Bio. Ecol., 330, 493–504, https://doi.org/10.1016/j.jembe.2005.09.011, 2006.
Harvey, J. B. J., Johnson, S. B., Fisher, J. L., Peterson, W. T., and Vrijenhoek, R. C.: Comparison of morphological and next generation DNA sequencing methods for assessing zooplankton assemblages, J. Exp. Mar. Biol. Ecol., 487, 113–126, https://doi.org/10.1016/j.jembe.2016.12.002, 2017.
Hattenrath-Lehmann, T. K. and Gobler, C. J.: Identification of unique microbiomes associated with harmful algal blooms caused by Alexandrium fundyense and Dinophysis acuminata, Harmful Algae, 68, 17–30, https://doi.org/10.1016/j.hal.2017.07.003, 2017.
He, X., McLean, J. S., Edlund, A., Yooseph, S., Hall, A. P., Liu, S. Y., Dorrestein, P. C., Esquenazi, E., Hunter, R. C., Cheng, G., Nelson, K. E., Lux, R., and Shi, W.: Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle, P. Natl. Acad. Sci. USA, 112, 244–249, https://doi.org/10.1073/pnas.1419038112, 2015.
Herring, S. C., Christidis, N., Hoell, A., Hoerling, M. P., and Stott, P. A.: Explaining Extreme Events of 2017 from a Climate Perspective, Bull. Am. Meteorol. Soc., 100, S1–S117, https://doi.org/10.1175/bams-explainingextremeevents2017.1, 2019.
Hird, S. M., Sánchez, C., Carstens, B. C., and Brumfield, R. T.: Comparative gut microbiota of 59 neotropical bird species, Front. Microbiol., 6, 1403, https://doi.org/10.3389/fmicb.2015.01403, 2015.
Hitchcock, J. N., Mitrovic, S. M., Hadwen, W. L., Roelke, D. L., Growns, I. O., and Rohlfs, A. M.: Terrestrial dissolved organic carbon subsidizes estuarine zooplankton: An in situ mesocosm study, Limnol. Oceanogr., 61, 254–267, https://doi.org/10.1002/lno.10207, 2016.
Hugenholtz, P., Tyson, G. W., Webb, R. I., Wagner, A. M., and Blackall, L. L.: Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria, with no known pure-culture representatives, Appl. Environ. Microbiol., 67, 411–419, https://doi.org/10.1128/AEM.67.1.411-419.2001, 2001.
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., Ruscheweyh, H. J., and Tappu, R.: MEGAN Community Edition – Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data, PLoS Comput. Biol., 12, 1–12, https://doi.org/10.1371/journal.pcbi.1004957, 2016.
Huyer, A., Knoll, M., Paluszkiewicz, T., and Smith, R. L.: The Peru Undercurrent: a study in variability, Deep-Sea Res. Pt. A, 38, S247–S271, https://doi.org/10.1016/s0198-0149(12)80012-4, 1991.
Ianora, A.: Copepod life history traits in subtemperate regions, J. Mar. Syst., 15, 337–349, https://doi.org/10.1016/S0924-7963(97)00085-7, 1998.
Pachauri, R. K., Allen, M. R., Barros, V. R., Broome, J., Cramer, W., Christ, R., Church, J. A., Clarke, L., Dahe, Q., Dasgupta, P., and Dubash, N. K.: Climate change 2014: synthesis report. Contribution of Working Groups I, II and III to the fifth assessment report of the Intergovernmental Panel on Climate Change, p. 151, IPCC, 2014.
Jaffe, A. L., Thomas, A. D., He, C., Keren, R., Valentin-Alvarado, L. E., Munk, P., Bouma-Gregson, K., Farag, I. F., Amano, Y., Sachdeva, R., West, P. T., and Banfield, J. F.: Patterns of gene content and co-occurrence constrain the evolutionary path toward animal association in Candidate Phyla Radiation Bacteria, MBio, 12, e00521-21, https://doi.org/10.1128/mBio.00521-21, 2021.
Kahru, M., Michell, B. G., Diaz, A., and Miura, M.: MODIS detects a devastating algal bloom in Paracas Bay, Peru, Eos, Trans. Am. Geophys. Union, 85, 465–472, 2004.
Kelly, R. P., Port, J. A., Yamahara, K. M., and Crowder, L. B.: Using environmental DNA to census marine fishes in a large mesocosm, PLoS One, 9, e86175, https://doi.org/10.1371/journal.pone.0086175, 2014.
Kim, S. and Park, M. G.: Paulinella longichromatophora sp. nov., a New Marine Photosynthetic Testate Amoeba Containing a Chromatophore, Protist, 167, 1–12, https://doi.org/10.1016/j.protis.2015.11.003, 2016.
Kolody, B. C., McCrow, J. P., Allen, L. Z., Aylward, F. O., Fontanez, K. M., Moustafa, A., Moniruzzaman, M., Chavez, F. P., Scholin, C. A., Allen, E. E., Worden, A. Z., Delong, E. F., and Allen, A. E.: Diel transcriptional response of a California Current plankton microbiome to light, low iron, and enduring viral infection, ISME J., 13, 2817–2833, https://doi.org/10.1038/s41396-019-0472-2, 2019.
Koumandou, V. L., Nisbet, R. E. R., Barbrook, A. C., and Howe, C. J.: Dinoflagellate chloroplasts – Where have all the genes gone?, Trends Genet., 20, 261–267, https://doi.org/10.1016/j.tig.2004.03.008, 2004.
Kudela, R. M., Lane, J. Q., and Cochlan, W. P.: The potential role of anthropogenically derived nitrogen in the growth of harmful algae in California, USA, Harmful Algae, 8, 103–110, https://doi.org/10.1016/j.hal.2008.08.019, 2008.
Kudela, R. M., Seeyave, S., and Cochlan, W. P.: The role of nutrients in regulation and promotion of harmful algal blooms in upwelling systems, Prog. Oceanogr., 85, 122–135, https://doi.org/10.1016/j.pocean.2010.02.008, 2010.
Kuehbacher, T., Rehman, A., Lepage, P., Hellmig, S., Fölsch, U. R., Schreiber, S., and Ott, S. J.: Intestinal TM7 bacterial phylogenies in active inflammatory bowel disease, J. Med. Microbiol., 57, 1569–1576, https://doi.org/10.1099/jmm.0.47719-0, 2008.
Lamb, J. S., Satgé, Y. G., and Jodice, P. G. R.: Diet composition and provisioning rates of nestlings determine reproductive success in a subtropical seabird, Mar. Ecol. Prog. Ser., 581, 149–164, https://doi.org/10.3354/meps12301, 2017.
Lemos, L. N., Medeiros, J. D., Dini-Andreote, F., Fernandes, G. R., Varani, A. M., Oliveira, G., and Pylro, V. S.: Genomic signatures and co-occurrence patterns of the ultra-small Saccharimonadia (phylum CPR/Patescibacteria) suggest a symbiotic lifestyle, Mol. Ecol., 28, 4259–4271, https://doi.org/10.1111/mec.15208, 2019.
Leray, M., Yang, J. Y., Meyer, C. P., Mills, S. C., Agudelo, N., Ranwez, V., Boehm, J. T., and Machida, R. J.: A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents, Front. Zool., 10, 1–14, https://doi.org/10.1186/1742-9994-10-34, 2013.
Limardo, A. J., Sudek, S., Choi, C. J., Poirier, C., Rii, Y. M., Blum, M., Roth, R., Goodenough, U., Church, M. J., and Worden, A. Z.: Quantitative biogeography of picoprasinophytes establishes ecotype distributions and significant contributions to marine phytoplankton, Environ. Microbiol., 19, 3219–3234, https://doi.org/10.1111/1462-2920.13812, 2017.
Lin, S., Zhang, H., Hou, Y., Zhuang, Y., and Miranda, L.: High-level diversity of dinoflagellates in the natural environment, revealed by assessment of mitochondrial cox1 and cob genes for dinoflagellate DNA barcoding, Appl. Environ. Microbiol., 75, 1279–1290, https://doi.org/10.1128/AEM.01578-08, 2009.
Marcy, Y., Ouverney, C., Bik, E. M., Lösekann, T., Ivanova, N., Martin, H. G., Szeto, E., Platt, D., Hugenholtz, P., Relman, D. A., and Quake, S. R.: Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth, P. Natl. Acad. Sci. USA, 104, 11889–11894, https://doi.org/10.1073/pnas.0704662104, 2007.
Margalef, R.: Life-forms of phytoplankton as survival alternatives in an unstable environment, Ocean. Acta, 1, 493–509, 1978.
Martin, J. L., Santi, I., Pitta, P., John, U., and Gypens, N.: Towards quantitative metabarcoding of eukaryotic plankton: an approach to improve 18S rRNA gene copy number bias, Metabarcod. Metagenom., 6, 245–259, https://doi.org/10.3897/mbmg.6.85794, 2022.
Martin, M.: Cutadapt removes adapter sequences from high-throughput sequencing reads, EMBnet J., 17, 10–12, https://doi.org/10.14806/ej.17.1.200, 2011.
Martino, C., Morton, J. T., Marotz, C. A., Thompson, L. R., Tripathi, A., Knight, R., and Zengler, K.: A Novel Sparse Compositional Technique Reveals Microbial Perturbations, MSystems, 4, e00016-19, https://doi.org/10.1128/msystems.00016-19, 2019.
Matsuyama, Y., Miyamoto, M., and Kotani, Y.: Grazing impacts of the heterotrophic dinoflagellate Polykrikos kofoidii on a bloom of Gymnodinium catenatum, Aquat. Microb. Ecol., 17, 91–98, https://doi.org/10.3354/ame017091, 1999.
Messié, M. and Chavez, F. P.: Seasonal regulation of primary production in eastern boundary upwelling systems, Prog. Oceanogr., 134, 1–18, https://doi.org/10.1016/j.pocean.2014.10.011, 2015.
Min, M. and Pitz, K.: MBARI-BOG/KOSMOS_eDNA_paper: Initial submission to Biogeosciences (v1.0), Zenodo [code and data set], https://doi.org/10.5281/zenodo.7255826, 2022.
Miya, M., Sato, Y., Fukunaga, T., Sado, T., Poulsen, J.Y., Sato, K., Minamoto, T., Yamamoto, S., Yamanaka, H., Araki, H., and Kondoh, M.: MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species, Roy. Soc. Open Sci., 2, 150088, https://doi.org/10.1098/rsos.150088, 2015.
Monuki, K., Barber, P. H., and Gold, Z.: eDNA captures depth partitioning in a kelp forest ecosystem, PLoS One, 16, 1–17, https://doi.org/10.1371/journal.pone.0253104, 2021.
Morris, A. W. and Riley, J. P.: The determination of nitrate in sea water, Anal. Chim. Acta, 29, 272–279, https://doi.org/10.1016/S0003-2670(00)88614-6, 1963.
Morris, R. M., Rappé, M. S., Connon, S. A., Vergin, K. L., Siebold, W. A., Carlson, C. A., and Giovannoni, S. J.: SAR11 clade dominates ocean surface bacterioplankton communities, Nature, 420, 806–810, https://doi.org/10.1038/nature01240, 2002.
Mullin, J. B. and Riley, J. P.: The colorimetric determination of silicate with special reference to sea and natural waters, Anal. Chim. Acta, 12, 162–176, https://doi.org/10.1016/S0003-2670(00)87825-3, 1955.
Needham, D. M. and Fuhrman, J. A.: Pronounced daily succession of phytoplankton, archaea and bacteria following a spring bloom, Nat. Microbiol., 1, 16005, https://doi.org/10.1038/nmicrobiol.2016.5, 2016.
Neufeld, J. D., Schäfer, H., Cox, M. J., Boden, R., McDonald, I. R., and Murrell, J. C.: Stable-isotope probing implicates Methylophaga spp and novel Gammaproteobacteria in marine methanol and methylamine metabolism, ISME J., 1, 480–491, https://doi.org/10.1038/ismej.2007.65, 2007.
O'donnell, J. L., Kelly, R. P., Lowell, N. C., and Port, J. A.: Indexed PCR primers induce template- Specific bias in Large-Scale DNA sequencing studies, PLoS One, 11, 1–11, https://doi.org/10.1371/journal.pone.0148698, 2016.
Ohki, K., Yamada, K., Kamiya, M., and Yoshikawa, S.: Morphological, phylogenetic and physiological studies of pico-cyanobacteria isolated from the halocline of a saline Meromictic Lake, Lake Suigetsu, Japan, Microbes Environ., 27, 171–178, https://doi.org/10.1264/jsme2.ME11329, 2012.
Page, F. C.: Marine gymnamoebae, Institute of Terrestrial Ecology, 60 pp., 1983.
Parada, A. E., Needham, D. M., and Fuhrman, J. A.: Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples, Environ. Microbiol., 18, 1403–1414, https://doi.org/10.1111/1462-2920.13023, 2016.
Park, M. G. and Kim, M.: Prey specificity and feeding of the thecate mixotrophic dinoflagellate fragilidium duplocampanaeforme, J. Phycol., 46, 424–432, https://doi.org/10.1111/j.1529-8817.2010.00824.x, 2010.
Park, S., Jung, Y. T., Park, J. M., and Yoon, J. H.: Pseudohongiella acticola sp. nov., a novel gammaproteobacterium isolated from seawater, and emended description of the genus Pseudohongiella, Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol., 106, 809–815, https://doi.org/10.1007/s10482-014-0250-0, 2014.
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., and Hugenholtz, P.: A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life, Nat. Biotechnol., 36, 996–1004, https://doi.org/10.1038/nbt.4229, 2018.
Partensky, F., Blanchot, J., and Vaulot, D.: Differential distribution and ecology of Prochlorococcus and Synechococcus in oceanic waters: A review, Bull. Inst. Ocean., 19, 457–475, 1999.
Patterson, D., Nygaard, K., Steinberg, G., and Turley, C.: Heterotrophic flagellates and other protists associated with oceanic detritus throughout the water column in the mid north atlantic, J. Mar. Biol. Assoc. United Kingdom, 73, 67–95, https://doi.org/10.1017/S0025315400032653, 1993.
Patti, B., Guisande, C., Vergara, A. R., Riveiro, I., Maneiro, I., Barreiro, A., Bonanno, A., Buscaino, G., Cuttitta, A., Basilone, G., and Mazzola, S.: Factors responsible for the differences in satellite-based chlorophyll a concentration between the major global upwelling areas, Estuar. Coast. Shelf Sci., 76, 775–786, https://doi.org/10.1016/j.ecss.2007.08.005, 2008.
Pennington, J. T., Mahoney, K. L., Kuwahara, V. S., Kolber, D. D., Calienes, R., and Chavez, F. P.: Primary production in the eastern tropical Pacific: A review, Prog. Oceanogr., 69, 285–317, https://doi.org/10.1016/j.pocean.2006.03.012, 2006.
Penven, P., Echevin, V., Pasapera, J., Colas, F., and Tam, J.: Average circulation, seasonal cycle, and mesoscale dynamics of the Peru Current System: A modeling approach, J. Geophys. Res. C, 110, 1–21, https://doi.org/10.1029/2005JC002945, 2005.
Pitz, K., Truelove, N., Nye, C., Michisaki, R. P., and Chavez, F.: Environmental DNA (eDNA) 12S Metabarcoding Illumina MiSeq NGS PCR Protocol (Touchdown), protocols.io, https://doi.org/10.17504/protocols.io.bcppivmn, 2020.
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., Peplies, J., and Glöckner, F. O.: The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools, Nucl. Acids Res., 41, 590–596, https://doi.org/10.1093/nar/gks1219, 2013.
R Core Team: R: A Language and Environment for Statistical Computing, Vienna, Austria, http://www.R-project.org (last access: 21 February 2023), 2019.
Riebesell, U., Bellerby, R. G. J., Grossart, H. P., and Thingstad, F.: Mesocosm CO2 perturbation studies: From organism to community level, Biogeosciences, 5, 1157–1164, https://doi.org/10.5194/bg-5-1157-2008, 2008.
Riebesell, U., Czerny, J., Von Bröckel, K., Boxhammer, T., Büdenbender, J., Deckelnick, M., Fischer, M., Hoffmann, D., Krug, S. A., Lentz, U., Ludwig, A., Muche, R., and Schulz, K. G.: Technical Note: A mobile sea-going mesocosm system – New opportunities for ocean change research, Biogeosciences, 10, 1835–1847, https://doi.org/10.5194/bg-10-1835-2013, 2013.
Riemann, L., Steward, G. F., and Azam, F.: Erratum: Dynamics of bacterial community composition and activity during a mesocosm diatom bloom, Appl. Environ. Microbiol., 66, 2282, https://doi.org/10.1128/AEM.66.5.2282-2282.2000, 2000.
Rimet, F., Chaumeil, P., Keck, F., Kermarrec, L., Vasselon, V., Kahlert, M., Franc, A., and Bouchez, A.: R-Syst::diatom: An open-access and curated barcode database for diatoms and freshwater monitoring, Database, 1–21, https://doi.org/10.1093/database/baw016, 2016.
Robertson, D. A.: Possible functions of surface structure and size in some planktonic eggs of marine fishes, New Zeal. J. Mar. Freshw. Res., 15, 147–153, 1981.
Sandaa, R. A., Gómez-Consarnau, L., Pinhassi, J., Riemann, L., Malits, A., Weinbauer, M. G., Gasol, J. M., and Thingstad, T. F.: Viral control of bacterial biodiversity – Evidence from a nutrient-enriched marine mesocosm experiment, Environ. Microbiol., 11, 2585–2597, https://doi.org/10.1111/j.1462-2920.2009.01983.x, 2009.
Sassoubre, L. M., Yamahara, K. M., Gardner, L. D., Block, B. A., and Boehm, A. B.: Quantification of Environmental DNA (eDNA) Shedding and Decay Rates for Three Marine Fish, Environ. Sci. Technol., 50, 10456–10464, https://doi.org/10.1021/acs.est.6b03114, 2016.
Schnell, I. B., Bohmann, K., and Gilbert, M. T. P.: Tag jumps illuminated – reducing sequence-to-sample misidentifications in metabarcoding studies, Mol. Ecol. Resour., 15, 1289–1303, https://doi.org/10.1111/1755-0998.12402, 2015.
Schoenle, A., Hohlfeld, M., Rosse, M., Filz, P., Wylezich, C., Nitsche, F., and Arndt, H.: Global comparison of bicosoecid Cafeteria-like flagellates from the deep ocean and surface waters, with reorganization of the family Cafeteriaceae, Eur. J. Protistol., 73, 125665, https://doi.org/10.1016/j.ejop.2019.125665, 2020.
Silva, A. and Oliva, M.: Revisión sobre aspectos biológicos y de cultivo del lenguado chileno (Paralichthys adspersus), Lat. Am. J. Aquat. Res., 38, 377–386, 2010.
Simmons, M. P., Sudek, S., Monier, A., Limardo, A. J., Jimenez, V., Perle, C. R., Elrod, V. A., Pennington, J. T., and Worden, A. Z.: Abundance and biogeography of picoprasinophyte ecotypes and other phytoplankton in the eastern North Pacific Ocean, Appl. Environ. Microbiol., 82, 1693–1705, https://doi.org/10.1128/AEM.02730-15, 2016.
Skjoldal, H. R., Wiebe, P. H., Postel, L., Knutsen, T., Kaartvedt, S., and Sameoto, D. D.: Intercomparison of zooplankton (net) sampling systems: Results from the ICES/GLOBEC sea-going workshop, Prog. Oceanogr., 108, 1–42, https://doi.org/10.1016/j.pocean.2012.10.006, 2013.
Smayda, T. J.: Adaptations and selection of harmful and other dinoflagellate species in upwelling systems. 2. Motility and migratory behaviour, Prog. Oceanogr., 85, 71–91, https://doi.org/10.1016/j.pocean.2010.02.005, 2010.
Smayda, T. J. and Trainer, V. L.: Dinoflagellate blooms in upwelling systems: Seeding, variability, and contrasts with diatom bloom behaviour, Prog. Oceanogr., 85, 92–107, https://doi.org/10.1016/j.pocean.2010.02.006, 2010.
Spear-bernstein, L. and Miller, K. R.: Unique Location of the Phycobiliprotein Light-Harvesting Pigment in the Cryptophyceae, J. Phycol., 25, 412–419, https://doi.org/10.1111/j.1529-8817.1989.tb00245.x, 1989.
Spilling, K., Olli, K., Lehtoranta, J., Kremp, A., Tedesco, L., Tamelander, T., Klais, R., Peltonen, H., and Tamminen, T.: Shifting diatom – dinoflagellate dominance during spring bloom in the Baltic Sea and its potential effects on biogeochemical cycling, Front. Mar. Sci., 5, 327, 2018.
Stewart, R. I. A., Dossena, M., Bohan, D. A., Jeppesen, E., Kordas, R. L., Ledger, M. E., Meerhoff, M., Moss, B., Mulder, C., Shurin, J. B., Suttle, B., Thompson, R., Trimmer, M., and Woodward, G.: Mesocosm Experiments as a Tool for Ecological Climate-Change Research, 1st Edn., Elsevier Ltd., 71–181, https://doi.org/10.1016/B978-0-12-417199-2.00002-1, 2013.
Stoeck, T., Bass, D., Nebel, M., Christen, R., Jones, M. D. M., Breiner, H. W., and Richards, T. A.: Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water, Mol. Ecol., 19, 21–31, https://doi.org/10.1111/j.1365-294X.2009.04480.x, 2010.
Sudek, S., Everroad, R. C., Gehman, A. L. M., Smith, J. M., Poirier, C. L., Chavez, F. P., and Worden, A. Z.: Cyanobacterial distributions along a physico-chemical gradient in the Northeastern Pacific Ocean, Environ. Microbiol., 17, 3692–3707, https://doi.org/10.1111/1462-2920.12742, 2015.
Suffrian, K., Simonelli, P., Nejstgaard, J. C., Putzeys, S., Carotenuto, Y., and Antia, A. N.: Microzooplankton grazing and phytoplankton growth in marine mesocosms with increased CO2 levels, Biogeosciences, 5, 1145–1156, https://doi.org/10.5194/bg-5-1145-2008, 2008.
Taberlet, P., Coissac, E., Hajibabaei, M., and Rieseberg, L.: Environmental DNA, Mol. Ecol., 21, 1789–1793, 2012.
Taucher, J., Bach, L. T., Boxhammer, T., Nauendorf, A., Achterberg, E. P., Algueró-Muñiz, M., Arístegui, J., Czerny, J., Esposito, M., Guan, W., Haunost, M., Horn, H. G., Ludwig, A., Meyer, J., Spisla, C., Sswat, M., Stange, P., Riebesell, U., Aberle-Malzahn, N., Archer, S., Boersma, M., Broda, N., Büdenbender, J., Clemmesen, C., Deckelnick, M., Dittmar, T., Dolores-Gelado, M., Dörner, I., Fernández-Urruzola, I., Fiedler, M., Fischer, M., Fritsche, P., Gomez, M., Grossart, H. P., Hattich, G., Hernández-Brito, J., Hernández-Hernández, N., Hernández-León, S., Hornick, T., Kolzenburg, R., Krebs, L., Kreuzburg, M., Lange, J. A. F., Lischka, S., Linsenbarth, S., Löscher, C., Martínez, I., Montoto, T., Nachtigall, K., Osma-Prado, N., Packard, T., Pansch, C., Posman, K., Ramírez-Bordón, B., Romero-Kutzner, V., Rummel, C., Salta, M., Martínez-Sánchez, I., Schröder, H., Sett, S., Singh, A., Suffrian, K., Tames-Espinosa, M., Voss, M., Walter, E., Wannicke, N., Xu, J., and Zark, M.: Influence of ocean acidification and deep water upwelling on oligotrophic plankton communities in the subtropical North Atlantic: Insights from an in situ mesocosm study, Front. Mar. Sci., 4, 85, https://doi.org/10.3389/fmars.2017.00085, 2017.
Tillmann, U.: Interactions between planktonic microalgae and protozoan grazers, J. Eukaryot. Microbiol., 51, 156–168, https://doi.org/10.1111/j.1550-7408.2004.tb00540.x, 2004.
Trainer, V. L., Pitcher, G. C., Reguera, B., and Smayda, T. J.: The distribution and impacts of harmful algal bloom species in eastern boundary upwelling systems, Prog. Oceanogr., 85, 33–52, https://doi.org/10.1016/j.pocean.2010.02.003, 2010.
Tyrrell, T. and Merico, A.: Emiliania huxleyi: bloom observations and the conditions that induce them, in: Coccolithophores, Springer Berlin, Heidelberg, 75–97, https://doi.org/10.1007/978-3-662-06278-4_4, 2004.
Ushio, M., Murata, K., Sado, T., Nishiumi, I., Takeshita, M., Iwasaki, W., and Miya, M.: Demonstration of the potential of environmental DNA as a tool for the detection of avian species, Sci. Rep., 8, 1–10, https://doi.org/10.1038/s41598-018-22817-5, 2018.
Valentini, A., Taberlet, P., Miaud, C., Civade, R., Herder, J., Thomsen, P. F., Bellemain, E., Besnard, A., Coissac, E., Boyer, F., Gaboriaud, C., Jean, P., Poulet, N., Roset, N., Copp, G. H., Geniez, P., Pont, D., Argillier, C., Baudoin, J. M., Peroux, T., Crivelli, A. J., Olivier, A., Acqueberge, M., Le Brun, M., Møller, P. R., Willerslev, E., and Dejean, T.: Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding, Mol. Ecol., 25, 929–942, https://doi.org/10.1111/mec.13428, 2016.
Vincent, F. J., Colin, S., Romac, S., Scalco, E., Bittner, L., Garcia, Y., Lopes, R. M., Dolan, J. R., Zingone, A., and De Vargas, C.: The epibiotic life of the cosmopolitan diatom Fragilariopsis doliolus on heterotrophic ciliates in the open ocean, ISME J., 12, 1094–1108, 2018.
Walz, K., Yamahara, K., Michisaki, R. P., and Chavez, F. P.: MBARI Environmental DNA (eDNA) extraction using Qiagen DNeasy Blood and Tissue Kit, protocols.io, https://doi.org/10.17504/protocols.io.xjufknw, 2019.
Wear, E. K., Wilbanks, E. G., Nelson, C. E., and Carlson, C. A.: Primer selection impacts specific population abundances but not community dynamics in a monthly time-series 16S rRNA gene amplicon analysis of coastal marine bacterioplankton, Environ. Microbiol., 20, 2709–2726, https://doi.org/10.1111/1462-2920.14091, 2018.
Wiebe, P. H. and Holland, W. R.: Plankton Patchiness: Effects on Repeated Net Tows, Limnol. Oceanogr., 13, 315–321, https://doi.org/10.4319/lo.1968.13.2.0315, 1968.
Worden, A. Z., Nolan, J. K., and Palenik, B.: Assessing the dynamics and ecology of marine picophytoplankton: The importance of the eukaryotic component, Limnol. Oceanogr., 49, 168–179, https://doi.org/10.4319/lo.2004.49.1.0168, 2004.
Xu, L., Wu, Y. H., Jian, S. L., Wang, C. S., Wu, M., Cheng, L., and Xu, X. W.: Pseudohongiella nitratireducens sp. Nov., isolated from seawater, and emended description of the genus Pseudohongiella, Int. J. Syst. Evol. Microbiol., 66, 5155–5160, https://doi.org/10.1099/ijsem.0.001489, 2016.
Yang, C., Li, Y., Zhou, Y., Zheng, W., Tian, Y., and Zheng, T.: Bacterial community dynamics during a bloom caused by Akashiwo sanguinea in the Xiamen sea area, China, Harmful Algae, 20, 132–141, https://doi.org/10.1016/j.hal.2012.09.002, 2012.
Yang, C., Li, Y., Zhou, B., Zhou, Y., Zheng, W., Tian, Y., Van Nostrand, J. D., Wu, L., He, Z., Zhou, J., and Zheng, T.: Illumina sequencing-based analysis of free-living bacterial community dynamics during an Akashiwo sanguine bloom in Xiamen sea, China, Sci. Rep., 5, 1–11, https://doi.org/10.1038/srep08476, 2015.
Zubkov, M. V.: Faster growth of the major prokaryotic versus eukaryotic CO2 fixers in the oligotrophic ocean, Nat. Commun., 5, 1–6, https://doi.org/10.1038/ncomms4776, 2014.