the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 26 Jan 2024
| 26 Jan 2024
Continental scientific drilling and microbiology: (extremely) low biomass in bedrock of central Sweden
George Westmeijer
Cristina Escudero
Claudia Bergin
Stephanie Turner
Magnus Ståhle
Maliheh Mehrshad
Prune Leroy
Moritz Buck
Pilar López-Hernández
Jens Kallmeyer
Ricardo Amils
Stefan Bertilsson
Mark Dopson
Scientific drilling expeditions offer a unique opportunity to characterize microbial communities in the subsurface that have long been isolated from the surface. With subsurface microbial biomass being low in general, biological contamination from the drilling fluid, sample processing, or molecular work is a major concern. To address this, characterization of the contaminant populations in the drilling fluid and negative extraction controls are essential for assessing and evaluating such sequencing data. Here, rock cores down to 2250 m depth, groundwater-bearing fractures, and the drilling fluid were sampled for DNA to characterize the microbial communities using a broad genomic approach. However, even after removing potential contaminant populations present in the drilling fluid, notorious contaminants were abundant and mainly affiliated with the bacterial order Burkholderiales. These contaminant microorganisms likely originated from the reagents used for isolating DNA despite stringent quality standards during the molecular work. The detection of strictly anaerobic sulfate reducers such as Candidatus Desulforudis audaxviator suggested the presence of autochthonous deep biosphere taxa in the sequenced libraries, yet these clades represented only a minor fraction of the sequence counts (< 0.1 %), hindering further ecological interpretations. The described methods and findings emphasize the importance of sequencing extraction controls and can support experimental design for future microbiological studies in conjunction with continental drilling operations.
- Article
(2461 KB) - Full-text XML
- BibTeX
- EndNote





The concept of the deep biosphere was postulated by Thomas Gold (Gold, 1992) as life in the oceanic and continental subsurface plus subglacial lakes, with the continental deep biosphere being estimated to consist of 2 to 6×1029 cells (Magnabosco et al., 2018). These cells are active, greatly influence surface life through biogeochemical cycling (Kieft, 2016; Amils et al., 2023), and include archaeal, bacterial, and eukaryotic populations (Borgonie et al., 2015; Bell et al., 2020, 2022) as well as viruses (Rahlff et al., 2021; Holmfeldt et al., 2021; Engelhardt et al., 2014). Together with infiltration from the photosynthesis-driven surface (Osterholz et al., 2022), geogenic hydrogen and carbon dioxide from the deep subsurface are the main energy sources for subsurface life (Pedersen, 2000). Research on the subsurface has been enabled and accelerated by the use of springs (Suzuki et al., 2014), artesian wells (Chapelle et al., 2002), boreholes (Bomberg et al., 2016), mines (Magnabosco et al., 2016), and dedicated underground laboratories (Boylan et al., 2019; Wu et al., 2016). Genomic approaches have greatly increased understanding of microbial diversity in the continental subsurface and revealed a large proportion of diversity for which the ecology is not fully understood (Hug et al., 2016).
Subsurface microorganisms inhabit groundwater-filled fractures (Wu et al., 2017), are attached to minerals on the rock surface (Casar et al., 2020), and reside in the pore space of rocks as endoliths (Escudero et al., 2018). The pore space in rocks provides these endolithic communities with a surface to colonize, mineral nutrients, and moisture (Golubic et al., 1981). Studies on endolithic communities emphasize the importance of depth (Magnabosco et al., 2018), availability of water and electron donors (Dai et al., 2021), and pore space (Dutta et al., 2018) for microbial biomass and activity. Many of these organisms are chemolithoautotrophs and obtain energy through the oxidation of inorganic compounds, such as sulfide, iron(II), and hydrogen (Lever et al., 2015b). The majority of lithotrophs capture carbon dioxide through the energy-demanding Calvin cycle, and this, combined with the often energy- and nutrient-limiting conditions in the subsurface, limits microbial growth (Ghosh and Dam, 2009). Examples of these organisms previously identified in the subsurface are Thiobacillus denitrificans, which combines the oxidation of sulfides with the reduction of nitrate (Beller et al., 2006; Lopez-Fernandez et al., 2023), or Acidovorax sp., which couples the oxidation of ferrous iron to the reduction of nitrate (Pantke et al., 2012; Escudero et al., 2020). Studying the diversity and abundance of these endolithic communities would benefit the understanding of the microbial ecology and biomass in the continental subsurface.
In general, biomass in the continental subsurface is low, and hence contamination during field sampling or sample processing is a major concern, especially when drill cores are employed to retrieve the samples, as allochthonous populations in the drilling fluid are introduced into the subsurface (Smith et al., 2000). With microbial biomass usually being several orders of magnitude lower in the subsurface compared to surface environments (Starnawski et al., 2017), even the infiltration of a small volume of drilling fluid would risk the sample being compromised and unusable for studying microbial diversity. Consequently, contaminant populations introduced along with the drilling fluid need to be identified, and the magnitude of any such contamination needs to be assessed prior to downstream ecological interpretations. These contamination control methods often involve the introduction of tracers in the drilling fluid such as fluorescent microspheres or perfluorocarbon tracers that can be used to assess drilling fluid infiltration into the rock core (Kallmeyer, 2017). Removal of such tracers from the drill core surface through flaming allows one to minimize contamination of the inner core (Lever et al., 2006). Tracer count of the core surface can be combined with cell counts of the drilling fluid to assess microbial contamination originating from the drilling fluid. Furthermore, during molecular analyses, the typically low biomass in the deep continental biosphere increases the risk of detecting notorious reagent contaminants that would cause erroneous conclusions about the nature of the microbial community (Sheik et al., 2018; Salter et al., 2014). Despite the concerns regarding contamination, studies on this vast biome are necessary for understanding the role of the deep subsurface microbiome in carbon and nutrient cycling and would contribute to the growing body of literature on the continental subsurface.
Integration of subsurface microbiology into the workings of the International Continental Scientific Drilling Program (ICDP) raised several questions (Mangelsdorf and Kallmeyer, 2010), including understanding subsurface biodiversity, interactions between the deep biosphere and geosphere, and deep life as an analog for potential life on other planets. Pursuing answers to these questions, two subsurface scientific drilling projects termed Collisional Orogeny in the Scandinavian Caledonides (COSC-1 and COSC-2), located in western central Sweden, were performed in 2014 and 2020 (Lorenz et al., 2015, 2022). Here, microorganisms present in a hard rock environment were studied down to 2250 m depth by sampling drill cores and groundwater-bearing fractures during the COSC-2 drilling project (IGSN: ICDP5054EH40001). The efficacy of the contamination control measures was evaluated by characterizing the microorganisms in the samples of interest combined with those present in the drilling fluid.
2.1 Rock core samples
The aim of this study was to characterize the microbial community in the pore space of subsurface rocks (n=50) and natural fractures (n=29) covering various rock types (Fig. 1). The drilling fluid was sampled () simultaneously with the rocks and natural fractures (see Sect. 2.2) to assess the efficiency of the decontamination methods. The drilling fluid contained on average 335±396 cells mL−1 (mean ± SD, n=44, range 1–1554 cells mL−1). Based on the fluorescent beads being extinguished during sample decontamination (Fig. 2), on average 98.9±3.3 % of these cells were estimated to be destroyed by flame sterilization of the core surface, and for 31 of the 44 cores, over 99.9 % of the cells were destroyed.
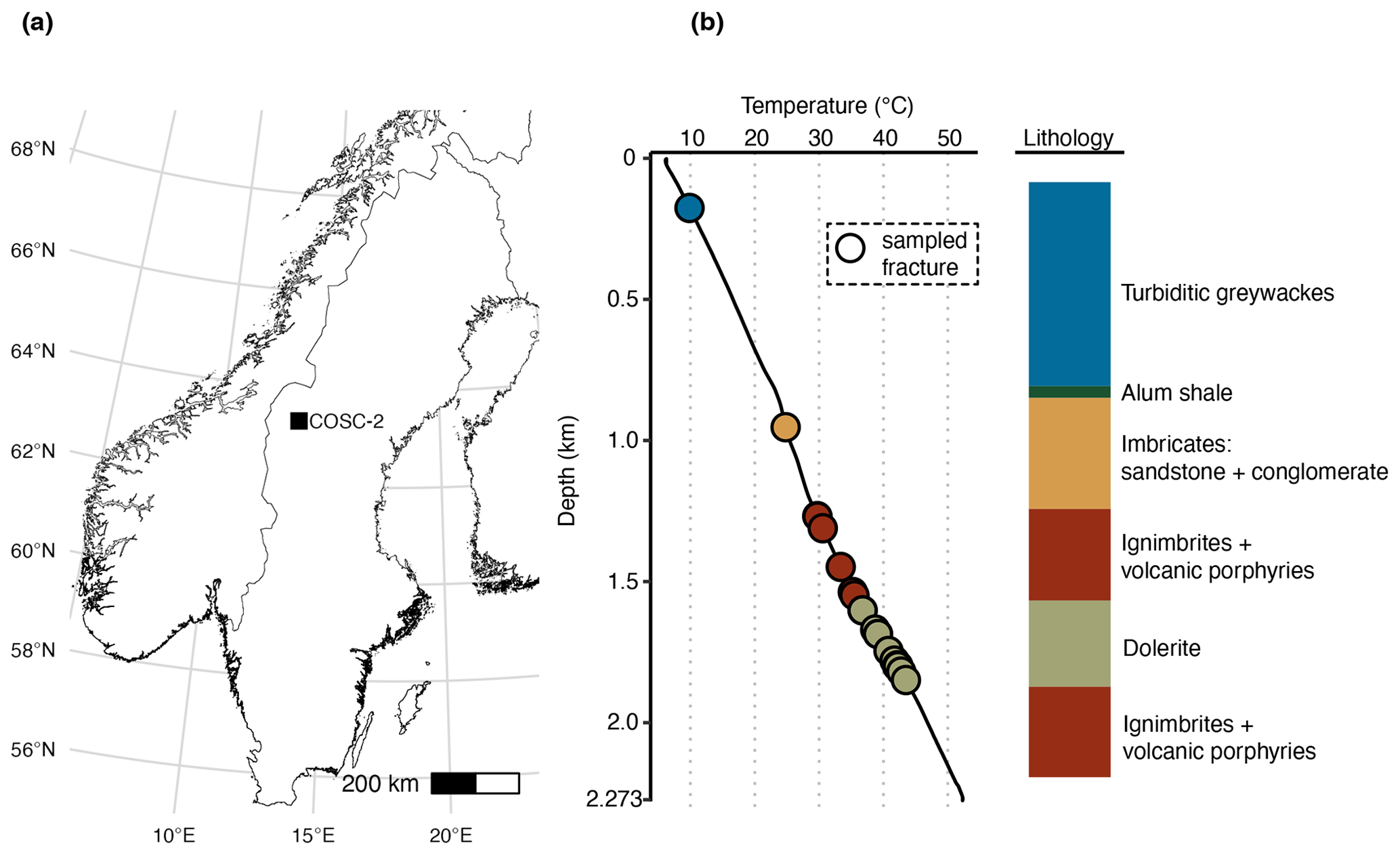
Figure 1Location and lithology sampling site. (a) The COSC-2 borehole within Sweden (lat 63.3124∘ N, long 13.5265∘ E). (b) Temperature profile and lithology according to depth. Points represent sampled natural fractures (n=29), colored according to lithology. Drill cores were sampled at 50 m intervals from 100 to 2250 m depth (n=50), covering the various lithologies. Temperature increased linearly with depth, ranging from 6.1 ∘C at the top to 52.3 ∘C at the bottom of the borehole. Temperature data from Lorenz et al. (2021).
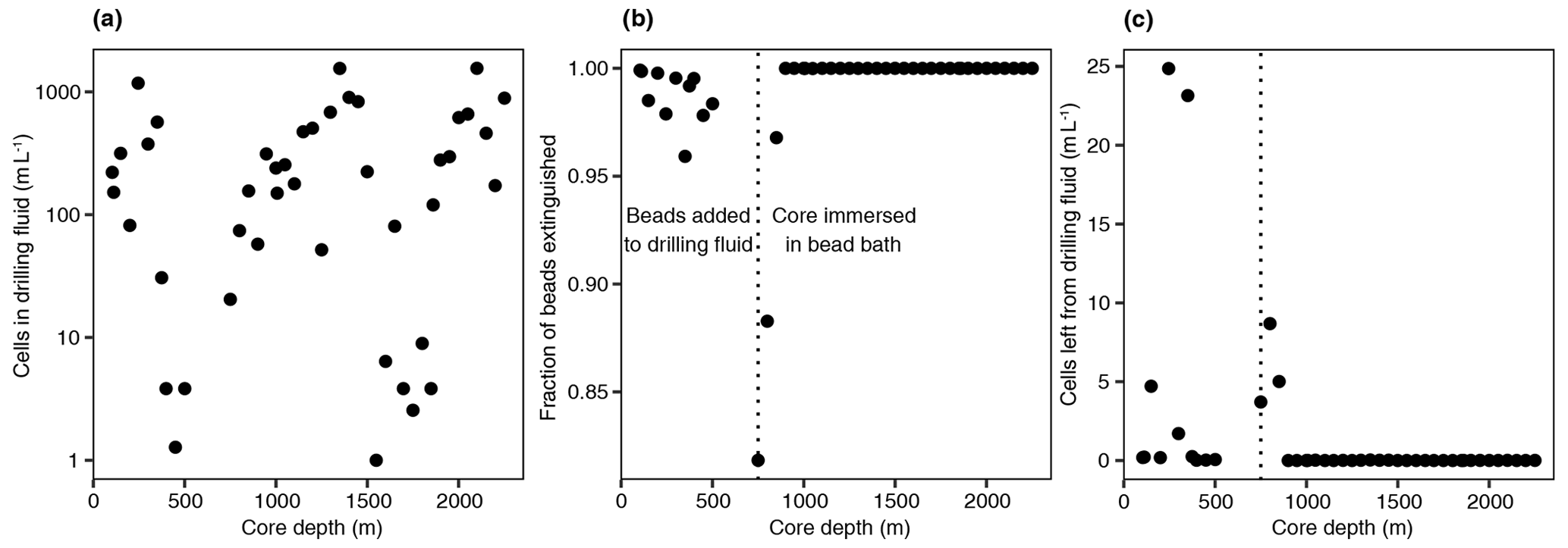
Figure 2Drilling fluid and contamination control. (a) Cell numbers in the drilling fluid over depth, quantified using fluorescence microscopy. (b) Fraction of extinguished fluorescent beads using flame sterilization of the core surface. The number of beads was determined before and after surface sterilization using fluorescence microscopy, and the higher the fraction of beads extinguished, the more successful the sterilization procedure. At 750 m depth, an alternative method of adding the beads was used by immersing the core in a bead bath instead of adding the beads to the drilling fluid due to adhesion of the beads to clay particles. (c) The estimated number of remaining beads after sterilization by multiplying the cell number in the drilling fluid (a) by the fraction of intact beads (b).
Biases are introduced during both DNA extraction and PCR amplification (Teske and Sørensen, 2008; Morono et al., 2014). To address this, six extraction protocols were tested using methods based on phenol-chloroform adapted to low-biomass rock samples and commercially available kits while maximizing input material (ground rock). To minimize amplification bias, three different primer pairs were tested, targeting both bacteria and archaea. Despite these measures, none of the extracts from the cores contained detectable DNA (<0.05 ng µL−1). This suggested that surface sterilization of the rock cores, to remove contaminants from the drilling fluid, was successful as the majority of the cells from the drilling fluid on the core surface were destroyed (Fig. 2) and DNA could be isolated (in detectable quantities) from the drilling fluid itself. Amplification of potentially extracted DNA, targeting different regions of the 16S ribosomal RNA gene using three primer pairs, also did not yield any products. Furthermore, whole genome amplification confirmed the extremely low concentration of DNA as qPCR of the amplified product revealed that, based on similar quantification cycles (Cq), the samples of interest and the negative extraction controls were indistinguishable. Whole genome amplification has previously been shown to amplify DNA from a single cell (Braslavsky et al., 2003) and has been successfully employed on crystalline rock samples (Puente-Sánchez et al., 2018). As previous studies have described microbes to persist within the deep bedrock (Puente-Sánchez et al., 2014, 2018) and crystalline bedrock of the Fennoscandian Shield (Purkamo et al., 2020), the lack of microbial cells in the rock samples scrutinized in this study underlines the influence of local lithology on microbial abundance with pore space, the presence of any electron donor, and the availability of water as potential controlling factors. The cause of the observed low biomass was likely a combination of these factors, especially given the variable lithology (Fig. 1). However, the effect of the lithology on the biomass was not feasible for prediction prior to field sampling as the expected lithology from the siting differed from the drilled lithology (Lorenz et al., 2022).
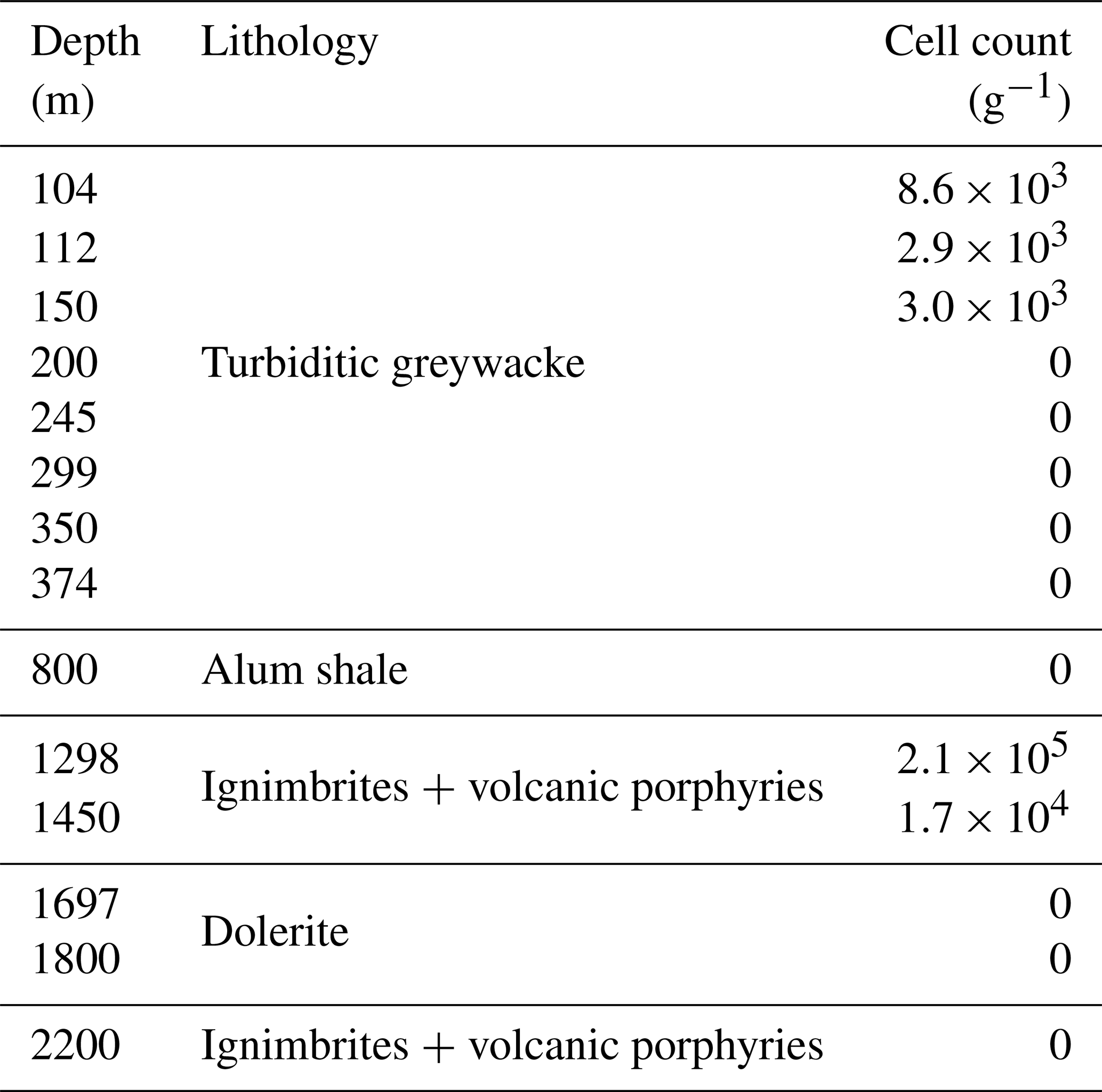
In addition to DNA extraction and amplification, cell numbers were determined using fluorescence microscopy. DNA staining was observed in the shallowest rock samples (100–150 m depth) and in the fault zone around 1300 m depth (Fig. 3; Table 1). However, the presence of microorganisms could not be confirmed by fluorescence in situ hybridization with catalyzed reporter deposition (CARD-FISH) as the corresponding fluorescent signals were deemed unspecific. The lack of a reliable signal could be due to (i) the extremely low abundance of microbial populations; (ii) a low number of ribosomes present in the cells, although this is unlikely given the signal amplification capacity of the CARD method; (iii) insufficient membrane permeabilization despite the use of standard permeabilization methods for Gram-positive bacteria and archaea; or (iv) the presence of microbial populations that fail to hybridize with the general bacterial (EUB338 I–III) and archaeal (ARC915) probes even though these probes have been reported as hybridizing with a high proportion of microbial diversity (Moter and Göbel, 2000). Given the low detection limit of the method (able to detect single cells; Hoshino et al., 2008) and its widespread application in low-biomass environments (Escudero et al., 2018; Teske, 2005), an extremely low microbial biomass was the most probable cause of the lack of a fluorescent signal.
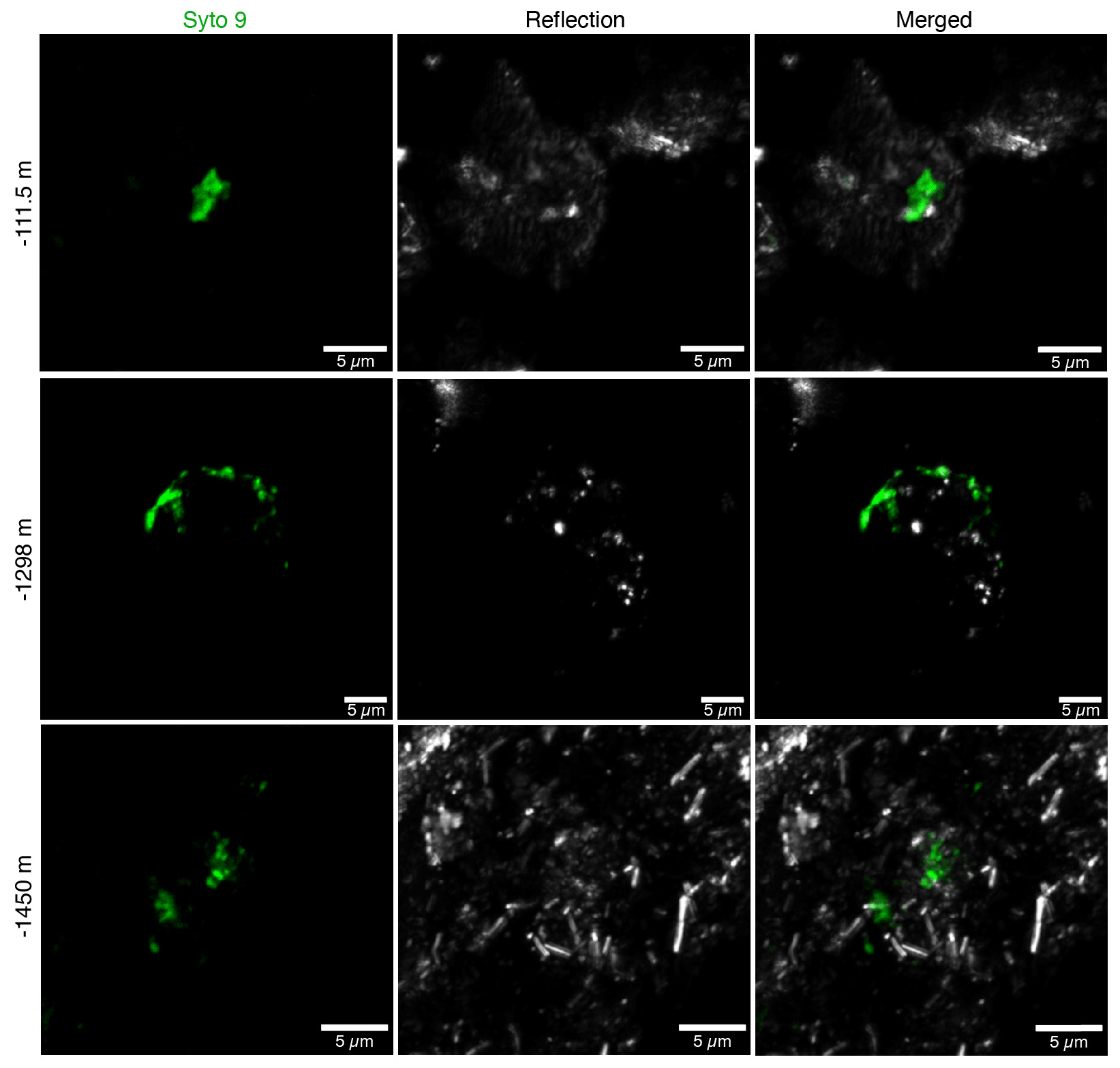
Figure 3Fluorescence micrographs of microorganisms detected in drill cores at 112, 1298, and 1450 m depth. Microbial populations were visualized with the DNA-binding dye Syto-9 (in green) with the reflection images in grey (mainly minerals). The presence of microorganisms could not be confirmed with CARD-FISH as the fluorescent signals were deemed unspecific.
Table 1Cell counts on selected drill cores. Microbial cells were quantified using fluorescence microscopy combined with the DNA-binding dyes DAPI and Syto-9. However, the presence of microorganisms could not be confirmed by CARD-FISH (described in the Results section).
2.2 Fracture samples
DNA was isolated from the groundwater-bearing fractures (n=7) occurring in a fault zone between 1200 and 1700 m depth and their respective drilling fluid samples (n=6). Sequencing of the 16S ribosomal RNA gene yielded 2758 unique amplicon sequence variants (ASVs). Bacterial 16S rRNA copy numbers were in the range of 103 to 105 copies per gram fracture sample, while archaea were hardly detected with copy numbers close to the lower detection limit of the qPCR (Table 2).
Table 2Details of fracture samples. Bacterial and archaeal abundances in the DNA extracts were quantified using qPCR combined with bacterial and archaeal primers targeting the 16S ribosomal RNA gene. The gene copies are presented as the average per-gram fracture sample ± standard deviation (SD) (n=6). The fractures at 1268 and 1270 m depth shared a drilling fluid sample as these fractures were part of the same drill core.
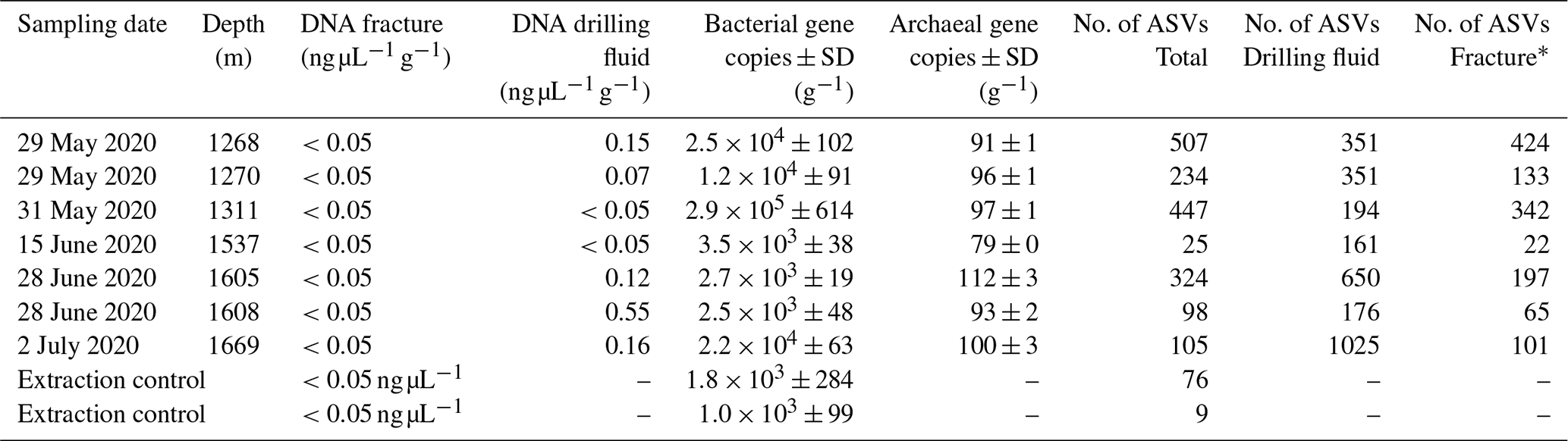
* Number of ASVs after removing the ASVs present in the drilling fluid.
The difference in composition between the microbial communities in the drilling fluid compared to the fractures was small but significant (permutational multivariate analysis of variance, MANOVA; R2=0.17; p=0.01). Removal of ASVs detected in the negative extraction controls (n=78) and the drilling fluid (n=1724) from the fracture samples reduced the sequence count in the latter by 52 % and 64 %, respectively (Fig. 4); 1304 ASVs were exclusively detected in the fracture samples, representing 11 % of the sequence counts, and after removing the contaminant populations from the fracture samples for downstream analysis, the difference between the latter and the drilling fluid increased (R2=0.32, p=0.002). However, the composition of the microbial community on the fracture surfaces (Fig. 4) strongly suggested contamination due to a high abundance of notorious reagent contaminants such as Ralstonia (36 % of the sequence counts), Agrobacterium (4.7 %), Sphingomonas (2.7 %), and Corynebacterium (2.2 %; Salter et al., 2014; Sheik et al., 2018). Apart from Corynebacterium, these genera were also detected in the negative extraction control, and together these four genera comprise 46 % of all the sequence counts. Removing contaminants based on taxonomy (e.g., genus) instead of ASVs would partially resolve this issue. However, this led to only 306 ASVs (3.7 % of the sequence counts) being retained compared to 1304 ASVs (11 %) when using ASVs. Either way, both approaches indicated that the dataset was predominantly comprised of contaminant clades and was unsuitable for ecological interpretations. The high abundance of contaminant populations was also reflected in the qPCR data, with the gene copies in the fractures ranging from 103 to 105 copies per gram, while the extraction controls contained 103 copies per gram, giving a ratio as low as 1 to 10 for six out of the seven samples. These contaminants, known to be notorious reagent contaminants, particularly in commercially available kits, were most likely introduced during DNA extraction despite strict measurements to prevent this. Most reagents used were of a high quality, including molecular-grade phenol-chloroform, ethanol, and co-precipitant, suggesting that the contaminants originated from either the column-based commercial kit used for purifying the DNA extract or the lysis solution, despite combining sterile filtering (0.1 µm pore size) with autoclaving of the latter. To better adapt the DNA extraction protocol to the microbial biomass in the sampled environment, it could be desirable to subsample material to be extracted in order to quantify cell density (for example, using fluorescence microscopy) prior to starting molecular work.
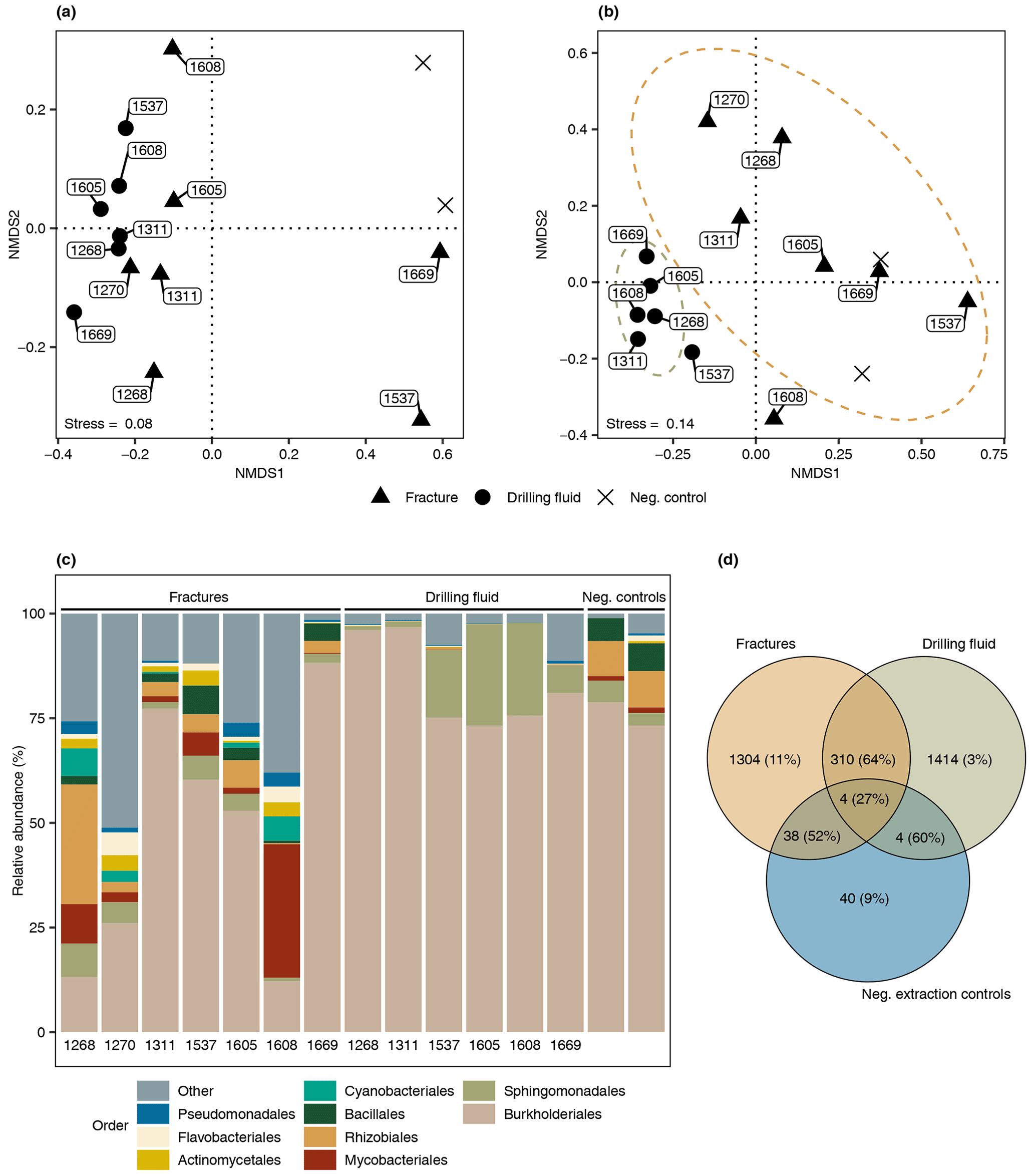
Figure 4Microbial community structure fracture samples. (a) Differentiation among microbial communities (beta diversity) prior to removing potential contaminants from the drilling fluid and the negative extraction controls. The labels depict the depth of the fracture in meters below the surface (elevation field site 320 m). Beta diversity was estimated according to Bray–Curtis dissimilarities and visualized using nonmetric dimensional scaling (NMDS). (b) Differentiation among microbial communities after removing ASVs in the drilling fluid from the respective fracture samples plus ASVs in the negative extraction controls. As shared ASVs between fracture and drilling fluid samples were excluded, the differentiation among these samples increased and, therefore, these samples were further apart. (c) Composition of the microbial community after removing potential contaminant ASVs from the fracture samples, depicting the nine most abundant orders with the remaining orders grouped as “Other”. The four most abundant orders were mainly represented by genera described as notorious reagent contaminants such as Ralstonia (Burkholderiales), Sphingomonas (Sphingomonadales), Corynebacterium (Mycobacteriales), and Agrobacterium (Rhizobiales). The text below each bar depicts the depth (m) of the fracture. (d) Overlap among the microbial communities in the fractures, drilling fluid, and negative extraction controls. The number of shared ASVs is followed by the abundance of these ASVs. For example, 38 ASVs were detected in both the fracture samples and the controls, and these taxa comprised 52 % of the sequence counts in the fracture samples.
Some taxa were more credible autochthonous populations as they are strict anaerobes and have been described in the deep subsurface, such as the sulfate reducers Pseudodesulfovibrio sp. or Candidatus Desulforudis audaxviator (Fig. 5). The latter has been reported in a South African gold mine at 2.8 km depth (Chivian et al., 2008), an aquifer at 2 km depth in Western Siberia (Karnachuk et al., 2019), and groundwater at 755 m depth in California (Becraft et al., 2021). However, as these taxa represent less than 0.1 % of the sequence counts, ecological interpretations of the data were hindered. In general, these observations emphasize the critical importance of negative extraction controls, in addition to characterization of the drilling fluid, to enable identification of contaminant populations.
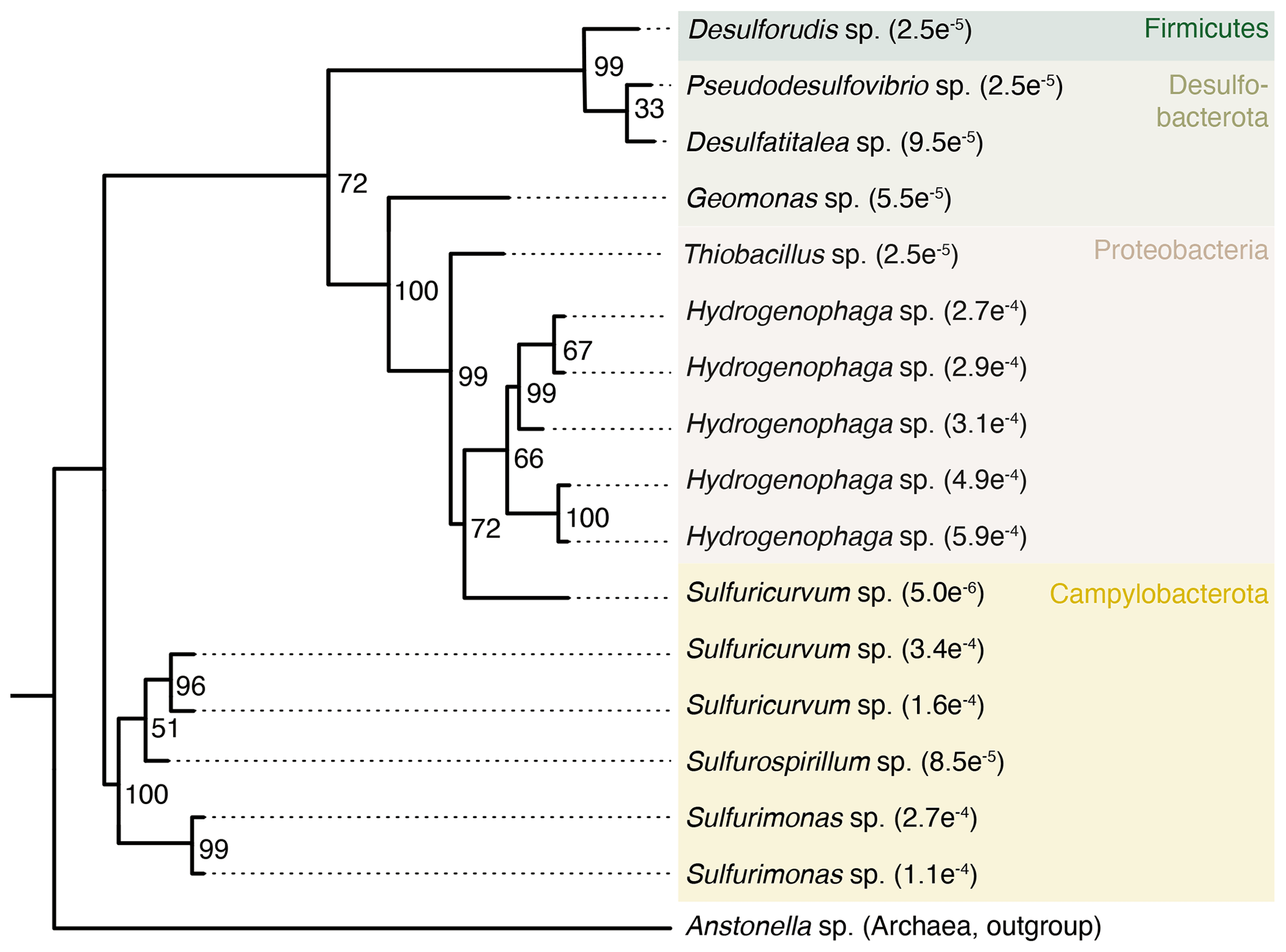
Figure 5Phylogenetic tree of selected ASVs in fracture samples. The tree was constructed using maximum likelihood on 16S rRNA gene amplicons (V3–V4 region, approximately 420 base pairs). Numbers on the nodes depict bootstrap values, with values of 100 and 70 indicating robust and moderate support, respectively. The number in the tip label shows the relative abundance with, for example, , showing that this ASV represents 0.027 % of all the sequence counts in the fracture samples. The ASVs included in the tree are unique to the fracture samples; i.e., they were not detected in the drilling fluid or in the extraction controls.
3.1 Site description
The COSC-2 borehole (lat 63.3124∘ N, long 13.5265∘ E; IGSN: ICDP5054EH40001) was drilled in an early Paleozoic rock formation with an elevation of 320 m and located in western central Sweden (Fig. 1). The top 780 m of the borehole was turbiditic greywacke that was followed by 45 m of sheared black shale. Underneath this shale zone, a sedimentary basement cover comprising sandstones and conglomerates extended downwards to 1250 m, followed by ignimbrites and volcanic porphyries down to the total depth of 2276 m, interrupted by dolerite intrusions between 1600 and 1930 m. Temperature increases linearly with depth with an increase of approximately 2 ∘C every 100 m, starting at 5 ∘C at the surface and rising to 55 ∘C at the maximum depth.
3.2 Field sampling
The sampling procedure and contamination control were described in detail in Lorenz et al. (2022). Briefly, rock core samples (n=50) were obtained from 100 to 2250 m depth at 50 m intervals. Samples were 20–30 cm in length and either 61 mm in diameter for 100–1550 m depth or 45 mm in diameter for 1600–2250 m depth. The drilling fluid was sampled simultaneously with core recovery to characterize potential contaminants introduced during the drilling operation. The collected drilling fluid was subsampled (1 mL) for cell counts, fixed with 2 % (vol vol) formaldehyde, stored in the dark at 4 ∘C, and transported to the home laboratory under identical conditions. Fluorescent microspheres (AFN-09, Radiant Color NV) were added to the drilling fluid at a concentration of 107–108 beads per milliliter (stock concentration 1018 beads per milliliter) that enabled us to assess the success of surface sterilization as the bead's fluorescence was extinguished when heated above 100 ∘C (Friese et al., 2017). Samples were collected and transported to the on-site microbiology laboratory, where the surface of the rock samples was briefly flame-sterilized to combust contaminants present in the drilling fluid, as evidenced by the destruction of the fluorescent microspheres. The number of beads on the core surface was counted using an on-site fluorescence microscope after sampling 2–3 g of surface fragments and compared to the bead concentration in the drilling fluid at the time of sampling. Surface sterilization was repeated until at least 99.9 % of the beads on the core surface could no longer be observed. At 750 m depth, an alternative strategy was utilized by immersing the cores in a bead bath (107–108 beads per milliliter) rather than by adding the beads to the drilling fluid due to adsorption of the fluorescent microspheres to clay particles. To do so, the bead concentration was quantified by washing the core's surface in a sterile plastic bag with 15 mL sterile, ultrapure water after immersing the core in the bead bath. This liquid was collected, brought onto a filter, stained, and analyzed with an on-site fluorescence microscope both before and after surface sterilization of the drill core. The core and the drilling fluid samples were stored on-site at −80 ∘C and transported on dry ice to the home laboratory, where they were kept at −80 ∘C until further processing.
In addition to the core samples, natural fractures were sampled (n=29) by collecting 2–3 g material surrounding the fracture using either flame-sterilized forceps or a chisel; 10 mL of sterile, ultrapure water was added to the material before storing the sample at −80 ∘C. Identical to the core samples, the drilling fluid was sampled simultaneously to sampling the fracture. Rock samples (2–3 g) for CARD-FISH were taken from the inner core, fixed in 4 % (vol vol) formaldehyde in Mackintosh minimal media, and stored on-site at 4 ∘C before being placed at −20 ∘C once back at the home laboratory.
3.3 Cell counts
Fluorescence microscopy was used to quantify the cells in the drilling fluid. To do so, 1× TE buffer (molecular grade, pH 8.0) was added to the fixed sample to a final volume of 10 mL, and this volume was brought onto a polycarbonate black membrane (pore size 0.22 µm, Whatman) at a pressure of 10–15 kPa and placed in a sterile Petri dish to dry. Meanwhile, a staining solution was prepared consisting of 30 % 100× SYBR Green I (Invitrogen), 10 % 10 g L−1 p-phenylenediamine (sterile-filtered, pore size 0.1 µm), and 60 % 1:1 glycerol in phosphate-buffered saline (PBS) (sterile-filtered, pore size 0.2 µm); 10 µL of the solution was brought onto a glass slide and cover slip before adding the filter and letting it incubate in the dark for 5 min. Finally, 300 cells or 10 fields of view (5×5 raster) were counted in the dark using a 100-fold objective on an epifluorescence microscope (Olympus BX50) combined with a light source (Olympus) that illuminated the DNA–dye complex at a wavelength of 497 nm.
3.4 CARD-FISH
Rock fragments were aseptically ground to sand grain size with a mortar and pestle. The ground rock was covered with 0.2 % (wt vol) agarose, dried at 37 ∘C, dehydrated with absolute ethanol, and stored at −20 ∘C. To account for autofluorescence and false positives, each sample was screened with fluorescence microscopy to select suitable fluorophores to avoid mineral interference. Additionally, controls were carried out using the Non338 probe combined with multiple fluorophores and DNA-binding dyes (DAPI and Syto-9) on sterilized rock fragments by acid wash and overnight incubation at 120 ∘C. More detail on the negative controls is provided in Escudero et al. (2018).
Hybridization, counter-staining, and mounting of the samples were performed as described in Pernthaler et al. (2008) using the probes EUB338 I–III (Amann et al., 1990; Daims et al., 1999), ARC915 (Stahl and Amann, 1991), and Non338 (Wallner et al., 1993). Endogenous peroxidases were inactivated using the protocol from Ishii et al. (2004). Samples were imaged using a confocal laser-scanning microscope (LSM710, Zeiss) coupled to an inverted platform (Axio Observer, Zeiss) and equipped with diode (405 nm), argon (458/488/514 nm), helium (543 nm), and neon lasers (633 nm). Images were collected using a 63-fold/1.4 oil immersion lens. Only the signals that matched the spectrum of the specific fluorophore were considered positive signals.
3.5 DNA extraction and amplification
Twelve rock samples covering all the lithologies (Fig. 1) were subsampled using a flame-sterilized hammer and chisel (ASC Scientific) on a metal benchtop that was sterilized with both bleach and 70 % ethanol. Subsamples were taken from the inner core, avoiding the outer 10–20 mm of the core to reduce potential contamination introduced during field sampling. Several subsamples were prepared simultaneously to avoid repeated freeze–thaw of the original sample. The rock fragments were ground using a flame-sterilized percussion mortar and pestle (ASC Scientific) and stored at −20 ∘C until DNA extraction. To verify whether the processing of the rock fragments did not hinder downstream work, a positive control was carried out by adding 109 cells of Pseudomonas aeruginosa to sterilized rock fragments prior to grinding the material, while flame-sterilized rock fragments served as a negative control. Extractions were performed in a room dedicated to extraction of nucleic acids while working in a fume hood that was sterilized with both bleach and 70 % ethanol. A maximum of eight samples (including controls) were extracted simultaneously, and the reagents were aliquoted according to the volume needed for processing the specific number of samples. For all the methods described, the reagents used were molecular grade, and all plastic consumables (tips, centrifuge tubes) were PCR grade and placed for 10 min in a UV hood before moving the material to the fume hood. Glassware was baked at 160 ∘C for 3 h, and the lysis solution was filter-sterilized (0.1 µm pore size) prior to autoclaving. The autoclave was cleaned prior to use and filled with fresh deionized water. After each extraction, the DNA yield was measured using a Qubit 2.0 fluorometer (Life Technologies) with a lower detection limit of 0.05 ng µL−1. As detectable amounts of DNA were not to be expected due to the low biomass, the DNA extracts subsequently served as a template in a PCR with 34 cycles using the primer pair 341F–805R (Herlemann et al., 2011, product length approximately 450 bp). To account for the potential fragmented nature of the DNA, PCR was also run using the bacterial primer pair 908F–1075R (Ohkuma and Kudo, 1998) covering a target region of approximately 170 bp. The archaeal primer pair 915F–1059R (Yu et al., 2005) was used to check for the potential presence of archaea. The three PCRs served as screening for potentially isolated DNA.
Initial attempts to extract DNA used the FastDNA Spin kit for soil (MP Biomedicals), DNeasy PowerSoil kit (Qiagen), and the DNeasy PowerMax Soil kit (Qiagen). The latter was attempted due to the large amount of input material (10 g). According to the manufacturer's instructions, the final elution volume was 5 mL, and this volume was concentrated to 50 µL using the DNA/RNA Clean All kit (Norgen Biotek). Due to potential adsorption of DNA to particles, an extraction protocol was designed according to Lever et al. (2015a) using 10 g of ground rock. Briefly, this protocol included a pyrophosphate wash (200 mM) prior to cell lysis (50 ∘C for 2 h), chemical lysis based on a lysis solution (pH 10) containing 800 mM guanidium-HCl, 30 mM ethylenediaminetetraacetic acid (EDTA), 0.5 % Triton X-100, and 30 mM Tris-HCl, 1 min of bead beating using the rock fragments as beads, purification using phenol–chloroform–isoamyl alcohol (pH 8.0) combined with phase-lock tubes (Qiagen), a chloroform–isoamyl alcohol wash, and precipitation overnight at −20 ∘C using ethanol NaCl combined with linear polyacrylamide as a co-precipitant. The obtained pellet was dried, washed with 70 % ethanol, dissolved in 100 µL nuclease-free water, and purified using the DNA/RNA Clean All kit (Norgen Biotek). Finally, the nucleic acids were eluted in 35 µL nuclease-free water and used as a template for whole genome amplification (described below). To increase the input material for DNA extraction even further, 100 g of ground rock from two cores was washed while shaking (300 rpm) with 250 mL 1 M phosphate buffer (pH 7) for 1 h at room temperature. The supernatant was decanted and filtered using a membrane filter (0.1 µm pore size, 45 mm in diameter) combined with a vacuum source to capture the cells. The membrane was directly subjected to DNA extraction using the DNeasy PowerWater kit (Qiagen), adjusting the elution volume to 50 µL. The negative extraction control was 250 mL of an identical buffer without any rock added and subjected to identical downstream processing.
Fracture samples (n=29) were extracted using the custom protocol based on Lever et al. (2015a) described above, adding the entire sample (2 to 3 g). The paired drilling fluid samples (n=28) were extracted by bringing 30 mL of the fluid onto a membrane filter (pore size 0.1 µm) using a vacuum source. The filter served as input for the DNeasy PowerWater kit (Qiagen), following the manufacturer's instructions except for eluting the nucleic acids in 50 µL instead of in 100 µL. The DNA concentrations of the fracture samples were all below the detection limit (0.05 ng µL−1), while the drilling fluid isolates contained 0.1–1.1 ng µL−1. PCR amplification (34 cycles, primer pair 341F–805R) resulted in a product for 7 out of 29 fracture samples. These amplified products and those from the respective drilling fluid samples were sequenced on an Illumina MiSeq platform at the Science for Life Laboratory, Sweden, generating 2×300 bp paired-end reads. Two negative extraction controls were subjected to identical processing, including amplification, and added to the sequencing project.
3.6 Whole genome amplification and PCR screening of amplified DNA
Rock DNA extracts were amplified with the Repli-g Single Cell kit (Qiagen) following the manufacturer's protocol with minor adaptations. DNA amplification was done in 384-well plates to allow for small reaction volumes and in 96-well plates for larger volumes. The plates were UV-treated with 2 J while covered with an optical sealing, and used the same day. Plastic consumables, lysis buffer, neutralization reagent, and nuclease-free water were UV-treated with 2 J directly prior to use and kept on ice. For 10 µL reaction volume, 1 µL of template was lysed with 0.5 µL D1 and neutralized with 0.5 µL N1. For 50 µL reaction volume, 2.5 µL template was lysed with 2.5 µL D1 and neutralized with 5 µL N1. Lysis was done at 95 ∘C for 15 s, followed by 5 min at room temperature before reactions were placed on ice; 0.5 µM SYTO 13 (Invitrogen) was added to the amplification mix as a reporter dye. The amplification was run at 30 ∘C for 8 h with fluorescence readings every 15 min on a qPCR machine (CFX384 C1000, Bio-Rad) or plate reader (FLUOStar Omega, BMG LABTECH). The reactions were inactivated at 65 ∘C for 5 min and then stored at −20 ∘C. The amplified DNA was diluted up to 1000-fold and screened with the 341F–805R primers targeting the bacterial 16S rRNA (Herlemann et al., 2011). The PCR was run with LightCycler 480 SYBR Green I Master mix (Roche), primers at a final concentration of 0.25 µM, and 2 µL of diluted amplified DNA in a reaction volume of 10 µL on a LightCycler 480 (Roche). Amplification settings were 3 min at 95 ∘C, 40 cycles of 15 s 95 ∘C, 30 s 57 ∘C, and 45 s at 72 ∘C, followed by a melting curve.
3.7 Real-time PCR
Microbial abundance was assessed by quantifying the 16S ribosomal RNA gene copies using real-time PCR (qPCR) on a LightCycler 480 (Roche Diagnostics). The reaction volume (10 µL) consisted of 5 µL Platinum SYBR Green qPCR SuperMix-UDG with ROX (Thermo Fisher Scientific), 0.4 µL 10 µM primer, 3.2 µL nuclease-free water, and 1.0 µL template. Bacterial and archaeal gene fragments were amplified using the primers 908F_mod/1075R (Ohkuma and Kudo, 1998) and 915F/1059R (Yu et al., 2005), respectively. Cycling conditions were 2 min at 95 ∘C, 40 cycles of 15 s at 95 ∘C, and 30 s at 60 ∘C, followed by a melt curve analysis to assess product specificity. Standard curves were generated with a dilution series of purified PCR product using genomic DNA of pure cultures as template (i.e., Acidiphilium cryptum JF-5 for bacteria and Ferroplasma acidiphilum BRGM4 for archaea). Standards, samples of interest, and no-template controls were run in triplicate, and the former two were 1:10 diluted in nuclease-free water to account for inhibition of the polymerase. The gene copy numbers were reported in gene copies per gram after correcting for the mass of the rock used for DNA extraction and the elution volume. Reaction efficiencies for the bacterial and archaeal assays were 95 % and 96 %, respectively. A minimum number of three quantification cycles (ΔCq) between samples and no-template controls were maintained as a limit of detection.
3.8 Bioinformatics and reproducibility
Raw sequencing reads from the 16S ribosomal RNA gene amplicons (n=13) were processed using the ampliseq pipeline (v2.3.0; Straub et al., 2020) from the nf-core framework that relied on Nextflow (v21.10.6), Cutadapt (v3.4), FastQC (v0.11.9), DADA2 (v1.22.0), and the SBDI Sativa curated 16S GTDB database (release 207; Lundin and Andersson, 2021). The ampliseq pipeline was run with default settings, except for the trimming of the primers, whereby reads not containing the primer sequence or containing double copies were discarded from downstream analysis. ASVs present in the drilling fluid were removed from their respective fracture sample. For example, the ASVs detected in the drilling fluid taken at 1605 m depth were removed from the fracture sampled at the same depth (Table 2). By doing so, 62 % of the total sequence counts were removed from the fracture samples. ASVs present in the negative extraction controls (n=76) were also excluded from downstream analysis. Absolute counts were standardized within a sample to relative abundances by dividing an ASV count by the total number of counts within a sample. The phylogenetic tree was constructed according to maximum likelihood while using the R libraries phangorn and ape (Paradis and Schliep, 2019; Schliep, 2011). The substitution model (GTR + G + I) was selected based on the Akaike information criterion. The phylogeny was evaluated by bootstrapping (n=1000). Statistics and data visualization were performed using R (v4.2.1).
The goal of this study was to characterize the subsurface microbial community in the pore space of rocks and natural fractures by extracting DNA to be used for constructing sequencing libraries combined with quantification of microbial abundance using CARD-FISH. The difficulty with extracting nucleic acids and the lack of a fluorescent signal in the CARD-FISH analysis both indicated an extremely low microbial biomass in the sampled drill cores. In contrast, DNA was isolated from groundwater-bearing fractures, although ecological interpretations of the microbial communities in these fractures were compromised due to a high abundance of notorious contaminant genera. These contaminant populations could not have been identified without sampling the drilling fluid and implementing negative extraction controls. The presence of strict anaerobic sulfate-reducing bacteria that have been described as autochthonous deep biosphere microorganisms suggested that subsurface microorganisms were present in the fractures. The method to trace contamination by adding fluorescent beads to the drilling fluid was evaluated as successful. Not only did it allow us to assess the success of surface sterilization, but it also allowed fluorescence microscopy on rock fragments taken from the inner core to quickly check whether any microspheres were present, indicating contamination. Finally, in addition to the drilling fluid, the source of the drilling fluid (in this case a nearby freshwater lake) could be sampled at the water inlet to better discriminate potential contaminant populations.
All sequencing data have been made publicly available at the European Nucleotide Archive under project reference PRJEB65120. The annotated ASVs, the number of reads throughout the bioinformatic workflow, and a compiled version of the R Markdown document are provided on GitHub at https://doi.org/10.5281/zenodo.10521982 (geweaa, 2024).
MD, SB, JK, and RA designed the research. GW, ST, MS, MM, PL, MB, PLH, SB, and MD performed field work. GW, CE, CB, and MS performed the molecular work. GW processed and performed ecological interpretations of the sequencing data. GW and MD wrote the manuscript with comments from all the authors. MD, SB, and RA provided funding. All the authors read and improved the final version.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors thank Henning Lorenz for coordinating the drilling project and his expertise during field sampling; the COSC-2 drilling team and the on-site scientific personnel, Adrian Martinez and Guillermo Mateos, for their assistance with the CARD-FISH experiments; and Remco Hoogervorst, Andreas Arleborg, Ezhilarasan Mani Ezhilan, and Egon and Ola Bertilsson for their assistance during field work.
The drilling was funded by the International Continental Scientific Drilling Project (ICDP; grant no. 4-2017) with co-funding for scientific operations provided by the Swedish Research Council (Vetenskapsrådet; grant nos. 2019-03688 and 2017-00642), the German Science Foundation (Deutsche Forschungsgemeinschaft DFG, grant no. SPP 1006), and the National Science Centre of Poland (Narodowym Centrum Nauki, project no. 2018/29/B/ST10/02315). The scientific drilling operations were performed using the national research infrastructure for scientific drilling, Riksriggen, funded by the Swedish Research Council. The study was supported by the Swedish Research Council (contract nos. 2018-04311 and 2017-04422). High-throughput sequencing was carried out at the National Genomics Infrastructure hosted by the Science for Life Laboratory, Sweden. Whole genome amplification and PCR screening were done at the SciLifeLab Microbial Single Cell Genomics Facility at Uppsala University. Stefan Bertilsson acknowledges financial support from the Swedish Research Council and Science for Life Laboratory. Bioinformatics analyses were carried out utilizing the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) at Uppsala University (grant nos. SNIC 2022/22-743 and SNIC 2022/6-242). The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at UPPMAX, Uppsala University, partially funded by the Swedish Research Council through grant agreement nos. 2022-06725 and 2018-05973.
This paper was edited by Tina Treude and reviewed by Rachel L. Harris and one anonymous referee.
Amann, R. I., Binder, B. J., Olson, R. J., Chisholm, S. W., Devereux, R., and Stahl, D. A.: Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations, Appl. Environ. Microbiol., 56, 1919–1925, https://doi.org/10.1128/aem.56.6.1919-1925.1990, 1990.
Amils, R., Escudero, C., Oggerin, M., Puente Sánchez, F., Arce Rodríguez, A., Fernández Remolar, D., Rodríguez, N., García Villadangos, M., Sanz, J. L., Briones, C., Sánchez-Román, M., Gómez, F., Leandro, T., Moreno-Paz, M., Prieto-Ballesteros, O., Molina, A., Tornos, F., Sánchez-Andrea, I., Timmis, K., Pieper, D. H., and Parro, V.: Coupled C, H, N, S and Fe biogeochemical cycles operating in the continental deep subsurface of the Iberian Pyrite Belt, Environ. Microbiol., 25, 428–453, https://doi.org/10.1111/1462-2920.16291, 2023.
Becraft, E. D., Lau Vetter, M. C. Y., Bezuidt, O. K. I., Brown, J. M., Labonté, J. M., Kauneckaite-Griguole, K., Salkauskaite, R., Alzbutas, G., Sackett, J. D., Kruger, B. R., Kadnikov, V., van Heerden, E., Moser, D., Ravin, N., Onstott, T., and Stepanauskas, R.: Evolutionary stasis of a deep subsurface microbial lineage, ISME J., 15, 2830–2842, https://doi.org/10.1038/s41396-021-00965-3, 2021.
Bell, E., Lamminmäki, T., Alneberg, J., Andersson, A. F., Qian, C., Xiong, W., Hettich, R. L., Frutschi, M., and Bernier-Latmani, R.: Active sulfur cycling in the terrestrial deep subsurface, ISME J., 14, 1260–1272, https://doi.org/10.1038/s41396-020-0602-x, 2020.
Bell, E., Lamminmäki, T., Alneberg, J., Qian, C., Xiong, W., Hettich, R. L., Frutschi, M., and Bernier-Latmani, R.: Active anaerobic methane oxidation and sulfur disproportionation in the deep terrestrial subsurface, ISME J., 16, 1583–1593, https://doi.org/10.1038/s41396-022-01207-w, 2022.
Beller, H. R., Chain, P. S. G., Letain, T. E., Chakicherla, A., Larimer, F. W., Richardson, P. M., Coleman, M. A., Wood, A. P., and Kelly, D. P.: The genome sequence of the obligately chemolithoautotrophic, facultatively anaerobic bacterium Thiobacillus denitrificans, J. Bacteriol., 188, 1473–1488, https://doi.org/10.1128/JB.188.4.1473-1488.2006, 2006.
Bomberg, M., Lamminmäki, T., and Itävaara, M.: Microbial communities and their predicted metabolic characteristics in deep fracture groundwaters of the crystalline bedrock at Olkiluoto, Finland, Biogeosciences, 13, 6031–6047, https://doi.org/10.5194/bg-13-6031-2016, 2016.
Borgonie, G., Linage-Alvarez, B., Ojo, A. O., Mundle, S. O. C., Freese, L. B., Van Rooyen, C., Kuloyo, O., Albertyn, J., Pohl, C., Cason, E. D., Vermeulen, J., Pienaar, C., Litthauer, D., Van Niekerk, H., Van Eeden, J., Lollar, B. S., Onstott, T. C., and Van Heerden, E.: Eukaryotic opportunists dominate the deep-subsurface biosphere in South Africa, Nat. Commun., 6, 8952, https://doi.org/10.1038/ncomms9952, 2015.
Boylan, A. A., Perez-Mon, C., Guillard, L., Burzan, N., Loreggian, L., Maisch, M., Kappler, A., Byrne, J. M., and Bernier-Latmani, R.: H2-fuelled microbial metabolism in Opalinus Clay, Appl. Clay Sci., 174, https://doi.org/10.1016/j.clay.2019.03.020, 2019.
Braslavsky, I., Hebert, B., Kartalov, E., and Quake, S. R.: Sequence information can be obtained from single DNA molecules, P. Natl. Acad. Sci. USA, 100, 3960–3964, https://doi.org/10.1073/pnas.0230489100, 2003.
Casar, C. P., Kruger, B. R., Flynn, T. M., Masterson, A. L., Momper, L. M., and Osburn, M. R.: Mineral-hosted biofilm communities in the continental deep subsurface, Deep Mine Microbial Observatory, SD, USA, Geobiology, 18, 508–522, https://doi.org/10.1111/gbi.12391, 2020.
Chapelle, F. H., O'Neill, K., Bradley, P. M., Methá, B. A., Ciufo, S. A., Knobel, L. R. L., and Lovley, D. R.: A hydrogen-based subsurface microbial community dominated by methanogens, Nature, 415, 312–315, https://doi.org/10.1038/415312a, 2002.
Chivian, D., Brodie, E. L., Alm, E. J., Culley, D. E., Dehal, P. S., DeSantis, T. Z., Gihring, T. M., Lapidus, A., Lin, L. H., Lowry, S. R., Moser, D. P., Richardson, P. M., Southam, G., Wanger, G., Pratt, L. M., Andersen, G. L., Hazen, T. C., Brockman, F. J., Arkin, A. P., and Onstott, T. C.: Environmental genomics reveals a single-species ecosystem deep within Earth, Science, 322, 275–278, https://doi.org/10.1126/science.1155495, 2008.
Dai, X., Wang, Y., Luo, L., Pfiffner, S. M., Li, G., Dong, Z., Xu, Z., Dong, H., and Huang, L.: Detection of the deep biosphere in metamorphic rocks from the Chinese continental scientific drilling, Geobiology, 19, 278–291, https://doi.org/10.1111/gbi.12430, 2021.
Daims, H., Brühl, A., Amann, R., Schleifer, K. H., and Wagner, M.: The domain-specific probe EUB338 is insufficient for the detection of all bacteria: Development and evaluation of a more comprehensive probe set, Syst. Appl. Microbiol., 22, 434–444, https://doi.org/10.1016/S0723-2020(99)80053-8, 1999.
Dutta, A., Dutta Gupta, S., Gupta, A., Sarkar, J., Roy, S., Mukherjee, A., and Sar, P.: Exploration of deep terrestrial subsurface microbiome in Late Cretaceous Deccan traps and underlying Archean basement, India, Sci. Rep.-UK, 8, 17459, https://doi.org/10.1038/s41598-018-35940-0, 2018.
Engelhardt, T., Kallmeyer, J., Cypionka, H., and Engelen, B.: High virus-to-cell ratios indicate ongoing production of viruses in deep subsurface sediments, ISME J., 8, 1538, https://doi.org/10.1038/ismej.2013.245, 2014.
Escudero, C., Vera, M., Oggerin, M., and Amils, R.: Active microbial biofilms in deep poor porous continental subsurface rocks, Sci. Rep.-UK, 8, 1538, https://doi.org/10.1038/s41598-018-19903-z, 2018.
Escudero, C., del Campo, A., Ares, J. R., Sánchez, C., Martínez, J. M., Gómez, F., and Amils, R.: Visualizing microorganism-mineral interaction in the Iberian Pyrite Belt subsurface: the Acidovorax case, Front. Microbiol., 11, 572104, https://doi.org/10.3389/fmicb.2020.572104, 2020.
Friese, A., Kallmeyer, J., Kitte, J. A., Martínez, I. M., Bijaksana, S., and Wagner, D.: A simple and inexpensive technique for assessing contamination during drilling operations, Limnol. Oceanogr.-Meth., 15, 200–211, https://doi.org/10.1002/lom3.10159, 2017.
geweaa: geweaa/fractures: Biogeosciences, Version v1.0, Zenodo [code], https://doi.org/10.5281/zenodo.10521982, 2024.
Ghosh, W. and Dam, B.: Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea, FEMS Microbiol. Rev., 33, 999–1043, https://doi.org/10.1111/j.1574-6976.2009.00187.x, 2009.
Gold, T.: The deep, hot biosphere, P. Natl. Acad. Sci. USA, 89, 6045–6049, https://doi.org/10.1073/pnas.89.13.6045, 1992.
Golubic, S., Friedmann, I., and Schneider, J.: The lithobiontic ecological niche, with special reference to microorganisms, J. Sediment. Petrol., 51, 475–478, https://doi.org/10.1306/212f7cb6-2b24-11d7-8648000102c1865d, 1981.
Herlemann, D. P. R., Labrenz, M., Jürgens, K., Bertilsson, S., Waniek, J. J., and Andersson, A. F.: Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea, ISME J., 5, 1571–1579, https://doi.org/10.1038/ismej.2011.41, 2011.
Holmfeldt, K., Nilsson, E., Simone, D., Lopez-Fernandez, M., Wu, X., de Bruijn, I., Lundin, D., Andersson, A. F., Bertilsson, S., and Dopson, M.: The Fennoscandian Shield deep terrestrial virosphere suggests slow motion 'boom and burst' cycles, Communications Biology, 4, 307, https://doi.org/10.1038/s42003-021-01810-1, 2021.
Hoshino, T., Yilmaz, L. S., Noguera, D. R., Daims, H., and Wagner, M.: Quantification of target molecules needed to detect microorganisms by fluorescence in situ hybridization (FISH) and catalyzed reporter deposition-FISH, Appl. Environ. Microb., 74, 5068-5077, https://doi.org/10.1128/AEM.00208-08, 2008.
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., Butterfield, C. N., Hernsdorf, A. W., Amano, Y., Ise, K., Suzuki, Y., Dudek, N., Relman, D. A., Finstad, K. M., Amundson, R., Thomas, B. C., and Banfield, J. F.: A new view of the tree of life, Nat. Microbiol., 1, 16048, https://doi.org/10.1038/nmicrobiol.2016.48, 2016.
Ishii, K., Mußmann, M., MacGregor, B. J., and Amann, R.: An improved fluorescence in situ hybridization protocol for the identification of bacteria and archaea in marine sediments, FEMS Microbiol. Ecol., 50, 203–213, https://doi.org/10.1016/j.femsec.2004.06.015, 2004.
Kallmeyer, J.: Contamination control for scientific drilling operations, Adv. Appl. Microbiol., 98, 61–91, https://doi.org/10.1016/bs.aambs.2016.09.003, 2017.
Karnachuk, O. V., Frank, Y. A., Lukina, A. P., Kadnikov, V. V., Beletsky, A. V., Mardanov, A. V., and Ravin, N. V.: Domestication of previously uncultivated Candidatus Desulforudis audaxviator from a deep aquifer in Siberia sheds light on its physiology and evolution, ISME J., 13, 1947–1959, https://doi.org/10.1038/s41396-019-0402-3, 2019.
Kieft, T. L.: Their world: a diversity of microbial environments, in: Microbiology of the deep continental biosphere, Springer, 225–249, https://doi.org/10.1007/978-3-319-28071-4_6, 2016.
Lever, M. A., Alperin, M., Engelen, B., Inagaki, F., Nakagawa, S., Steinsbu, B. O., and Teske, A.: Trends in basalt and sediment core contamination during IODP expedition 301, Geomicrobiol. J., 23, 517–530, https://doi.org/10.1080/01490450600897245, 2006.
Lever, M. A., Torti, A., Eickenbusch, P., Michaud, A. B., Šantl-Temkiv, T., and Jørgensen, B. B.: A modular method for the extraction of DNA and RNA, and the separation of DNA pools from diverse environmental sample types, Front. Microbiol., 6, 476, https://doi.org/10.3389/fmicb.2015.00476, 2015a.
Lever, M. A., Rogers, K. L., Lloyd, K. G., Overmann, J., Schink, B., Thauer, R. K., Hoehler, T. M., and Jørgensen, B. B.: Life under extreme energy limitation: A synthesis of laboratory- and field-based investigations, FEMS Microbiol. Rev., 39, 688–728, https://doi.org/10.1093/femsre/fuv020, 2015b.
Lopez-Fernandez, M., Westmeijer, G., Turner, S., Broman, E., Ståhle, M., Bertilsson, S., and Dopson, M.: Thiobacillus as a key player for biofilm formation in oligotrophic groundwaters of the Fennoscandian Shield, npj Biofilms Microbiomes, 9, 41, https://doi.org/10.1038/s41522-023-00408-1, 2023.
Lorenz, H., Rosberg, J.-E., Juhlin, C., Bjelm, L., Almqvist, B. S. G., Berthet, T., Conze, R., Gee, D. G., Klonowska, I., Pascal, C., Pedersen, K., Roberts, N. M. W., and Tsang, C.-F.: COSC-1 – drilling of a subduction-related allochthon in the Palaeozoic Caledonide orogen of Scandinavia, Sci. Dril., 19, 1–11, https://doi.org/10.5194/sd-19-1-2015, 2015.
Lorenz, H., Juhlin, C., Rosberg, J.-E., Bazargan, M., Klonowska, I., Kück, J., Lescoutre, R., Rejkjær, S., Westmeijer, G., and Ziemniak, G.: COSC-2 operational report – Operational data sets, GFZ Data Services [data set], https://doi.org/10.5880/ICDP.5054.003, 2021.
Lorenz, H., Rosberg, J.-E., Juhlin, C., Klonowska, I., Lescoutre, R., Westmeijer, G., Almqvist, B. S. G., Anderson, M., Bertilsson, S., Dopson, M., Kallmeyer, J., Kück, J., Lehnert, O., Menegon, L., Pascal, C., Rejkjær, S., and Roberts, N. N. W.: COSC-2 – drilling the basal décollement and underlying margin of palaeocontinent Baltica in the Paleozoic Caledonide Orogen of Scandinavia, Sci. Dril., 30, 43–57, https://doi.org/10.5194/sd-30-43-2022, 2022.
Lundin, D. and Andersson, A.: SBDI Sativa curated 16S GTDB database, SciLifeLab [data set], https://doi.org/10.17044/scilifelab.14869077.v4, 2021.
Magnabosco, C., Ryan, K., Lau, M. C. Y., Kuloyo, O., Sherwood Lollar, B., Kieft, T. L., Van HeerDen, E., and Onstott, T. C.: A metagenomic window into carbon metabolism at 3 km depth in Precambrian continental crust, ISME J., 10, 730–741, https://doi.org/10.1038/ismej.2015.150, 2016.
Magnabosco, C., Lin, L. H., Dong, H., Bomberg, M., Ghiorse, W., Stan-Lotter, H., Pedersen, K., Kieft, T. L., van Heerden, E., and Onstott, T. C.: The biomass and biodiversity of the continental subsurface, Nat. Geosci., 11, 707–717, https://doi.org/10.1038/s41561-018-0221-6, 2018.
Mangelsdorf, K. and Kallmeyer, J.: Integration of Deep Biosphere Research into the International Continental Scientific Drilling Program, Sci. Dril., 10, 46–55, https://doi.org/10.2204/iodp.sd.10.0.2010, 2010.
Morono, Y., Terada, T., Hoshino, T., and Inagaki, F.: Hot-alkaline DNA extraction method for deep-subseafloor archaeal communities, Appl. Environ. Microbiol., 80, 1985–1994, https://doi.org/10.1128/AEM.04150-13, 2014.
Moter, A. and Göbel, U. B.: Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms, J. Microbiol. Meth., 41, 85–112, https://doi.org/10.1016/S0167-7012(00)00152-4, 2000.
Ohkuma, M. and Kudo, T.: Phylogenetic analysis of the symbiotic intestinal microflora of the termite Cryptotermes domesticus, FEMS Microbiol. Lett., 164, 389–395, https://doi.org/10.1016/S0378-1097(98)00244-4, 1998.
Osterholz, H., Turner, S., Alakangas, L. J., Tullborg, E.-L., Dittmar, T., Kalinowski, B. E., and Dopson, M.: Terrigenous dissolved organic matter persists in the energy-limited deep groundwaters of the Fennoscandian Shield, Nat. Commun., 13, 4837, https://doi.org/10.1038/s41467-022-32457-z, 2022.
Pantke, C., Obst, M., Benzerara, K., Morin, G., Ona-Nguema, G., Dippon, U., and Kappler, A.: Green rust formation during Fe(II) oxidation by the nitrate-reducing Acidovorax sp. strain BoFeN1, Environ. Sci. Technol., 46, 1439–1446, https://doi.org/10.1021/es2016457, 2012.
Paradis, E. and Schliep, K.: ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R, Bioinformatics, 35, 526–528, https://doi.org/10.1093/bioinformatics/bty633, 2019.
Pedersen, K.: Exploration of deep intraterrestrial microbial life: current perspectives, FEMS Microbiol. Lett., 185, 9–16, https://doi.org/10.1111/j.1574-6968.2000.tb09033.x, 2000.
Pernthaler, A., Pernthaler, J., and Amann, R.: Sensitive multi-color fluorescence in situ hybridization for the identification of environmental microorganisms, in: Molecular Microbial Ecology Manual, edited by: Kowalchuk, G. A., de Bruijn, F. J., Head, I. M., Akkermans, A. D. L., and van Elsas, J. D., Kluwer Academic Publishers, 711–726, ISBN: 1402021763, 2008.
Puente-Sánchez, F., Moreno-Paz, M., Rivas, L. A., Cruz-Gil, P., García-Villadangos, M., Gómez, M. J., Postigo, M., Garrido, P., González-Toril, E., Briones, C., Fernández-Remolar, D., Stoker, C., Amils, R., and Parro, V.: Deep subsurface sulfate reduction and methanogenesis in the Iberian Pyrite Belt revealed through geochemistry and molecular biomarkers, Geobiology, 12, 34–47, https://doi.org/10.1111/gbi.12065, 2014.
Puente-Sánchez, F., Arce-Rodríguez, A., Oggerin, M., García-Villadangos, M., Moreno-Paz, M., Blanco, Y., Rodríguez, N., Bird, L., Lincoln, S. A., Tornos, F., Prieto-Ballesteros, O., Freeman, K. H., Pieper, D. H., Timmis, K. N., Amils, R., and Parro, V.: Viable cyanobacteria in the deep continental subsurface, P. Natl. Acad. Sci. USA, 115, 10702–10707, https://doi.org/10.1073/pnas.1808176115, 2018.
Purkamo, L., Kietäväinen, R., Nuppunen-Puputti, M., Bomberg, M., and Cousins, C.: Ultradeep microbial communities at 4.4 km within crystalline bedrock: Implications for habitability in a planetary context, Life, 10, 2, https://doi.org/10.3390/life10010002, 2020.
Rahlff, J., Turzynski, V., Esser, S. P., Monsees, I., Bornemann, T. L. V., Figueroa-Gonzalez, P. A., Schulz, F., Woyke, T., Klingl, A., Moraru, C., and Probst, A. J.: Lytic archaeal viruses infect abundant primary producers in Earth's crust, Nat. Commun., 12, 4642, https://doi.org/10.1038/s41467-021-24803-4, 2021.
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., Turner, P., Parkhill, J., Loman, N. J., and Walker, A. W.: Reagent and laboratory contamination can critically impact sequence-based microbiome analyses, BMC Biol., 12, 1–12, https://doi.org/10.1186/s12915-014-0087-z, 2014.
Schliep, K. P.: phangorn: phylogenetic analysis in R, Bioinformatics, 27, 592–593, https://doi.org/10.1093/bioinformatics/btq706, 2011.
Sheik, C. S., Reese, B. K., Twing, K. I., Sylvan, J. B., Grim, S. L., Schrenk, M. O., Sogin, M. L., and Colwell, F. S.: Identification and removal of contaminant sequences from ribosomal gene databases: Lessons from the Census of Deep Life, Front. Microbiol., 9, 840, https://doi.org/10.3389/fmicb.2018.00840, 2018.
Smith, D. C., Spivack, A. J., Fisk, M. R., Haveman, S. A., and Staudigel, H.: Tracer-based estimates of drilling-induced microbial contamination of deep sea crust, Geomicrobiol. J., 17, 207–219, https://doi.org/10.1080/01490450050121170, 2000.
Stahl, D. A. and Amann, R.: Development and application of nucleic acid probes in bacterial systematics, in: Nucleic acid techniques in bacterial systematics, edited by: Stackebrandt, E. and Goodfellow, M., John Wiley & Sons, Chichester, England, 205–248, ISBN 9780471929062, 1991.
Starnawski, P., Bataillon, T., Ettema, T. J. G., Jochum, L. M., Schreiber, L., Chen, X., Lever, M. A., Polz, M. F., Jørgensen, B. B., Schramm, A., and Kjeldsen, K. U.: Microbial community assembly and evolution in subseafloor sediment, P. Natl. Acad. Sci. USA, 114, 2940–2945, https://doi.org/10.1073/pnas.1614190114, 2017.
Straub, D., Blackwell, N., Langarica-Fuentes, A., Peltzer, A., Nahnsen, S., and Kleindienst, S.: Interpretations of environmental microbial community studies are biased by the selected 16S rRNA (gene) amplicon sequencing pipeline, Front. Microbiol., 11, 550420, https://doi.org/10.3389/fmicb.2020.550420, 2020.
Suzuki, Y., Konno, U., Fukuda, A., Komatsu, D. D., Hirota, A., Watanabe, K., Togo, Y., Morikawa, N., Hagiwara, H., Aosai, D., Iwatsuki, T., Tsunogai, U., Nagao, S., Ito, K., and Mizuno, T.: Biogeochemical signals from deep microbial life in terrestrial crust, PLoS One, 9, e113063, https://doi.org/10.1371/journal.pone.0113063, 2014.
Teske, A. and Sørensen, K. B.: Uncultured archaea in deep marine subsurface sediments: Have we caught them all?, ISME J., 2, 3–18, https://doi.org/10.1038/ismej.2007.90, 2008.
Teske, A. P.: The deep subsurface biosphere is alive and well, Trends Microbiol., 13, 402–404, https://doi.org/10.1016/j.tim.2005.07.004, 2005.
Wallner, G., Amann, R., and Beisker, W.: Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms, Cytometry, 14, 136–142, https://doi.org/10.1002/cyto.990140205, 1993.
Wu, X., Holmfeldt, K., Hubalek, V., Lundin, D., Åström, M., Bertilsson, S., and Dopson, M.: Microbial metagenomes from three aquifers in the Fennoscandian Shield terrestrial deep biosphere reveal metabolic partitioning among populations, ISME J., 10, 1192–1203, https://doi.org/10.1038/ismej.2015.185, 2016.
Wu, X., Pedersen, K., Edlund, J., Eriksson, L., Åström, M., Andersson, A. F., Bertilsson, S., and Dopson, M.: Potential for hydrogen-oxidizing chemolithoautotrophic and diazotrophic populations to initiate biofilm formation in oligotrophic, deep terrestrial subsurface waters, Microbiome, 5, 1–13, https://doi.org/10.1186/S40168-017-0253-Y, 2017.
Yu, Y., Lee, C., and Hwang, S.: Analysis of community structures in anaerobic processes using a quantitative real-time PCR method, Water Sci. Technol., 52, 85–91, https://doi.org/10.2166/wst.2005.0502, 2005.