the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Nov 2018
| 02 Nov 2018
Dynamic mercury methylation and demethylation in oligotrophic marine water
Kathleen M. Munson
Carl H. Lamborg
Rene M. Boiteau
Mak A. Saito
Mercury bioaccumulation in open-ocean food webs depends on the net rate of inorganic mercury conversion to monomethylmercury in the water column. We measured significant methylation rates across large gradients in oxygen utilization in the oligotrophic central Pacific Ocean. Overall, methylation rates over 24 h incubation periods were comparable to those previously published from Arctic and Mediterranean waters despite differences in productivity between these marine environments. In contrast to previous studies that have attributed Hg methylation to heterotrophic bacteria, we measured higher methylation rates in filtered water compared to unfiltered water. Furthermore, we observed enhanced demethylation of newly produced methylated mercury in incubations of unfiltered water relative to filtered water. The addition of station-specific bulk filtered particulate matter, a source of inorganic mercury substrate and other possibly influential compounds, did not stimulate sustained methylation, although transient enhancement of methylation occurred within 8 h of addition. The addition of dissolved inorganic cobalt also produced dramatic, if transient, increases in mercury methylation. Our results suggest important roles for noncellular or extracellular methylation mechanisms and demethylation in determining methylated mercury concentrations in marine oligotrophic waters. Methylation and demethylation occur dynamically in the open-ocean water column, even in regions with low accumulation of methylated mercury.
- Article
(2438 KB) - Full-text XML
-
Supplement
(1614 KB) - BibTeX
- EndNote





Mercury (Hg) is a toxic metal present in small amounts in the ocean that accumulates in fish to reach levels that pose human and environmental health risks. Because methylated Hg (collectively abbreviated as MeHg) accumulates more efficiently than inorganic Hg, (Hg(II)), transformations into MeHg ultimately control burdens in upper-trophic-level biota. These methylated species, monomethylmercury (MMHg) and dimethylmercury (DMHg), can be found throughout the water column, with concentration maxima often found at depths of rapid oxygen consumption (Mason and Fitzgerald, 1993; Cossa et al., 2009, 2011; Sunderland et al; 2009; Bowman et al., 2015, 2016). At these depths, production of methylated Hg has been linked to active organic matter remineralization (Cossa et al., 2009; Sunderland et al., 2009). Water column measurements of apparent Hg methylation rates in the Canadian Arctic Archipelago and the Mediterranean have attributed water column methylation to microbial processes (Lehnherr et al., 2011; Monperrus et al., 2007). However, the mechanism of methylation in these marine systems has not been determined.
Studies of obligate anaerobes from anoxic sediments have shown that Hg methylation is encoded by a pair of prokaryotic genes, hgcA and hgcB (Parks et al., 2013), the identification of which has revealed novel potential environments of methylation, such as sea ice (Gionfriddo et al., 2016), and methylation capacity across a greater diversity of microbial species (Gilmour et al., 2013, 2018). Analysis of microbial metagenomes from our 8∘ N (150–800 m) and the equatorial (50 m) stations identified sequences similar to microbial hgcA sequences encoding methylation but containing base pair substitutions, the effects of which on methylation potential are unknown (Podar et al., 2015). These findings, combined with a lack of determination of hgcAB distributions in oxic marine waters to date, demonstrate that the contribution of microbially mediated methylation pathways, such as hgcAB, to inventories of MeHg in oxic marine waters is still unknown.
We measured Pacific water column Hg methylation and MeHg demethylation rates using tracer-level stable isotope additions to examine potential sources of the MeHg maxima in low-oxygen waters. We anticipated that both methylation rates and demethylation rates would differ between waters with generally low MeHg concentrations found at the chlorophyll a maximum depths and the generally elevated MeHg concentrations found at the oxygen-minimum depths. We also hypothesized that MeHg concentrations in low-oxygen waters could be limited by organic matter supply. Thus, we expected that the addition of organic matter would promote net methylation. Alternatively, if methylation is limited by microbial activity, we expected that the stimulation of remineralization by carbon addition could enhance net methylation.
In order to gain insight into the mechanism of Hg methylation in the marine water column, we tested these hypotheses across gradients in concentrations of MeHg, total dissolved Hg (THg), dissolved oxygen, and apparent oxygen utilization (AOU) (Munson et al., 2015) in waters from the central Pacific Ocean collected between Hawaii (17∘ N) and American Samoa (12∘ S) during the METZYME cruise. Across this region, low-oxygen intermediate waters centered at 12∘ N set apart regions of higher MeHg concentrations (∼260 fM) in intermediate waters (Sunderland et al., 2009) from regions of low (< 100 fM) concentrations in the central and south Pacific, with the exception of relatively elevated concentrations centered around the Equator (Munson et al., 2015). The cruise track included regions of the North Pacific where intermediate waters incorporate anthropogenic Hg emissions (Sunderland et al., 2009; Lamborg et al., 2014) and marine fish Hg content has increased in recent decades (Drevnick et al., 2015).
Stock solutions of isotope-enriched spikes (MMHg: ∼ 225 µM; Hg: ∼500 µM) were prepared from isotopically enriched HgO (Oak Ridge National Laboratory) and stored at 4 ∘C until use. Isotopically enriched MM198Hg, for methylation rate measurements, and MM199Hg, used as an internal standard during analysis, were prepared from 198HgO and 199HgO, respectively, by methylation with methylcobalamin (Hintelmann and Ogrinc, 2003). A mass of 100 µg of enriched HgO was dissolved in 10 µL of HCl (conc, J. T. Baker) and diluted in 500 µL of sodium acetate buffer (0.1 M, pH 5). A mass of 500 µg of methylcobalamin (Sigma) was dissolved in 500 µL sodium acetate buffer and added to the Hg solution. The reaction proceeded for 3 h at room temperature before being stopped with 200 µL of KBr (0.3 M KBr in 2 M H2SO4). The enriched MMHg was extracted with toluene (400 µL, 3×). Extracts were combined and dried over sodium sulfate. A 100 µL aliquot of this primary stock was dissolved in 10 mL of isopropanol to produce a secondary stock solution. Carryover of methylcobalamin as Co in the stock solutions was found to be minimal (< 1 pM) from inductively coupled plasma mass spectrometry (ICPMS) analysis (Thermo Element 2).
Water for incubation experiments was collected onboard the R/V Kilo Moana between 1 and 24 October 2011 (Table 1). Water was collected from two depths at each station: (1) the chlorophyll a maximum and (2) the oxygen minimum identified from the Sea-Bird CTD package data on prior water sampling casts at each station (Table 1). Water was collected in acid-washed Niskin-X bottles deployed on a dedicated trace metal sampling rosette using AmSteel metal-free line. Water filtration, decanting, and treatment additions were performed under positive pressure from HEPA filter laminar flow hoods within clean bubbles constructed from plastic sheeting. Concentrations of dissolved Hg species (Table 1) were measured throughout the cruise transect (Munson et al., 2015), with the exception of 17∘ N, where no MMHg samples were preserved. Estimates of MMHg at the 17∘ N station incubation depths were made by subtracting MM200Hg contributions in isotope spike additions from the MeHg concentrations measured in initial time point incubation bottles.
Table 1Water column characteristics for Pacific Ocean waters from which potential mercury methylation rates were measured.
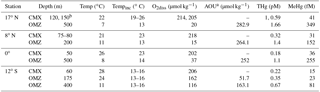
a AOU is a poor proxy for oxygen utilization in waters above 100 m due to gas exchange and photosynthesis. Values are therefore omitted for these depths. b Water was combined due to water budget limitations and water was a 1:3 mixture from 120 and 150 m in depth. c MMHg concentrations were not measured directly and were calculated from ambient Me200Hg in t0 incubation bottles by subtracting MMHg spike contributions.
Incubation water was decanted under N2 pressure either unfiltered or filtered through a 0.2 µm capsule filter (47 mm, Supor polyethersulfone membrane, Pall Corporation) into 20 L acid-washed polycarbonate mixing carboys (Nalgene). The carboys were covered in dark plastic bags during incubation setup to minimize exposure to light. Although hard to quantify, cells have been found in marine waters after filtration through 0.2 µm filters in the Mediterranean and Sargasso seas (Li and Dickie, 1985; Haller et al., 1999) under vacuum filtration. As a result, we cannot exclude the possibility that small cells, such as the SAR11 clade (Rappé et al., 2002), viruses, and cell material passed through filters and therefore were present in our filtered incubations. However, 0.2 µm filtration would potentially collect a subset of intact cells such as SAR11 due to clogging of filters and decreases in actual filter pore size. In addition, if the cells that pass through filters are dormant or, like SAR11, have slow growth rates (0.40—0.58 d−1) (Rappé et al., 2002), filtered waters would likely have lower cell density and less metabolic activity compared to unfiltered waters.
Pre-equilibrated spikes of isotopically enriched MM198Hg and 202Hg(II) were prepared by adding concentrated 202Hg(II) and MM198Hg to 0.2 µm filtered seawater and equilibrating at 16 ∘C in the dark for 24 h prior to use. Spikes were added using gas-tight syringes dedicated for use in isotope enrichment experiments (Hamilton).
Incubations were performed in triplicate in 250 mL amber borosilicate glass bottles (I-Chem) filled to the shoulder from mixing carboys leaving approximately 25 mL of headspace. As a result, in situ redox conditions were not maintained during incubation. After water addition, pre-equilibrated spikes of 202Hg(II) (396 fmol) and MM198Hg (126 fmol) were added. Thus, spike concentrations were added at approximately 1–14× ambient Hg(II) and 1–34× ambient MMHg concentrations. Treatments of carbon (1 mM, as succinate), inorganic cobalt (500 pM), and filtered particulate matter collected from McLane in situ pumps (Munson et al., 2015) were added to triplicate bottles. Pump filters were subsampled using a 2 cm (ID) acid-cleaned polycarbonate tube with a beveled edge. The 2 cm subsamples (punches) were cut in half using ceramic scissors. One-half of a subsample was added to each sample bottle for particulate amendments.
Bottles for the initial time point of incubation (t0) were preserved after isotope spike and treatment addition with 1 mL (0.5 %, final) of H2SO4 (conc., Fisher TM grade) and were stored at −4 ∘C until analysis. Bottles from stations at 17 and 8∘ N, and the Equator (0∘) were incubated in the dark for 24 h at temperatures maintained in refrigerators adjusted up to their highest temperature settings to most closely match in situ temperatures (Table 1). The station 12∘ S bottles were incubated in the dark up to 36 h in a time course study. After incubation, bottles for all time points were preserved with H2SO4 (0.5 %, final) and stored at −4 ∘C. Because DMHg decomposes to MMHg in acidic conditions (Black et al., 2009), our analysis measured the sum of DMHg and MMHg present in each bottle at the time of acidification. A previous study of methylation rate measurements in waters from Arctic Canada distinguished between production of MMHg and DMHg from Hg(II) as well as the conversion of MMHg to DMHg (Lehnherr et al., 2011). Rate constants of methylation to DMHg from Hg(II) were measured to be between 0.03 % and 1.22 % of those of MMHg production from Hg(II) (Lehnherr et al., 2011). However, in the same study, conversion of MMHg to DMHg proceeded more rapidly and rate constants ranged between 3.17 % and 43.3 % of those measured for MMHg production from Hg(II) (Lehnherr et al., 2011). In contrast, the rates presented here represent MMHg production from Hg(II) substrate and include any MMHg subsequently converted to DMHg.
Samples were analyzed as for MMHg using ascorbic-assisted direct ethylation (Munson et al., 2014), which generates volatile methylethylmercury from the sample analyte. Analysis of MMHg and Hg(II) concentrations and isotopic composition was performed on a ThermoFinnagan Element 2 ICPMS in the Plasma Mass Spectrometry Facility at the Woods Hole Oceanographic Institution linked to a Tekran 2700 automated methyl mercury detector (Tekran, Ontario, Canada). An internal MM199Hg standard (25 fmol) was added to samples and equilibrated for 24 h prior to MMHg determination to account for differences in ethylation efficiency among samples.
Hg isotopes were measured individually and relative counts were integrated using MATLAB scripts (provided in the Supplement) to quantify isotopic signals in the MMHg (as methylethylHg) and Hg(II) (as diethylHg) peaks. Resulting methylation of 202Hg(II) and demethylation of MM198Hg were quantified using a matrix inversion approach to solve the system of equations (Hintelmann and Ogrinc, 2003). Methylation rates were calculated from the observed ingrowth of Me202Hg divided by 202Hg(II) substrate supply and incubation time. Significant Me202Hg production was observed in t0 incubation bottles. As a result, the reported methylation rates were not calculated from the difference between final and initial time points (e.g., t24 h minus t0) as this often yielded a negative value indicating demethylation of Me202Hg between time points. Previous reports of methylation rates have differed in their considerations of available Hg(II) substrate, including assuming a constant supply of Hg(II) substrate (Monperrus et al., 2007) and an exponential model of decreasing Hg(II) substrate availability throughout the course of an incubation (Lehnherr et al., 2011). Generalized loss of Hg(II) was not an accurate representation of Hg(II) availability in our experiments. Although not analytically identical to total Hg measured by tin chloride reduction followed by cold vapor atomic fluorescence spectroscopy (CVAFS) analysis (Lamborg et al., 2012), we observed quantitative recovery of Hg(II) from the diethylHg peak of the Tekran 2700 instrument used for CVAFS analysis (p values < 0.05 from daily standard curves). Due to the presence of Hg(II) in the laboratory water used for reagent preparation, averages of Hg(II) peak integrations from reagent blanks (five to seven daily) were subtracted from those used to calculate daily standard curves. Sequential additions of reagents to filtered seawater (1×, 2×, and 3× reagent volume) did not change Hg(II) peak integrations after baseline correction. Results are presented as percentages of 202Hg(II) substrate that was methylated in each incubation bottle. Apparent methylation rate constants, km, were calculated as the percentage of 202Hg(II) substrate methylated over the incubation time.
For stations 17∘ N, 8∘ N, and 0∘, demethylation rates were determined from differences between measured MM198Hg at incubation time points (t24 minus t0) divided by the initial MM198Hg addition (126 fmol) and incubation time. At station 12∘ S, demethylation rates of both added MM198Hg spike and newly produced Me202Hg were measured from the time course experiments. These rates were calculated assuming first-order kinetics from the fit of exponential decay of the methylated Hg form versus incubation time (Prism 7, GraphPad).
Consistent with previous reports of methylation and demethylation rates (Monperrus et al., 2007; Lehnherr et al., 2011), our reported rates are potential rates and assume identical chemical behavior of added Hg(II) and MMHg isotopes to those species naturally present in the samples.
3.1 Unamended samples
Following 24 h incubation periods at the 17 and 8∘ N, and equatorial stations, we calculated first-order methylation and demethylation rates. Methylation rates were enhanced in 0.2 µm filtered water relative to unfiltered water (Fig. 1) with the exception of low-oxygen waters at the equatorial station. The observation of methylation in filtered seawater was unexpected given recent focus on the role of bacterial pathways analogous to those that occur in anoxic sediments (Monperrus et al., 2007; Lehnherr et al., 2011). However, abiotic mechanisms of methylation have been identified from experiments conducted in distilled water and nonmarine natural waters (Yin et al., 2014; Celo et al., 2006). Because the incubation waters we collected were influenced by the biota present prior to filtration, we do not distinguish between truly abiotic and biologically influenced mechanisms that occurred within these waters. Furthermore, although filtration may not remove all cells (Li and Dickie, 1985; Haller et al., 1999) we do not attribute the enhanced methylation observed in filtered waters to cellular mechanisms. Such an assumption would require a number of processes, such as reactivation of dormant cells, rapid rates of cell growth, and/or preferential selection of Hg(II) methylating cells during filtration. Instead, we assume that filtration removed the majority of heterotrophic bacteria relative to unfiltered waters therefore decreasing the likelihood of microbially mediated methylation in filtered samples.
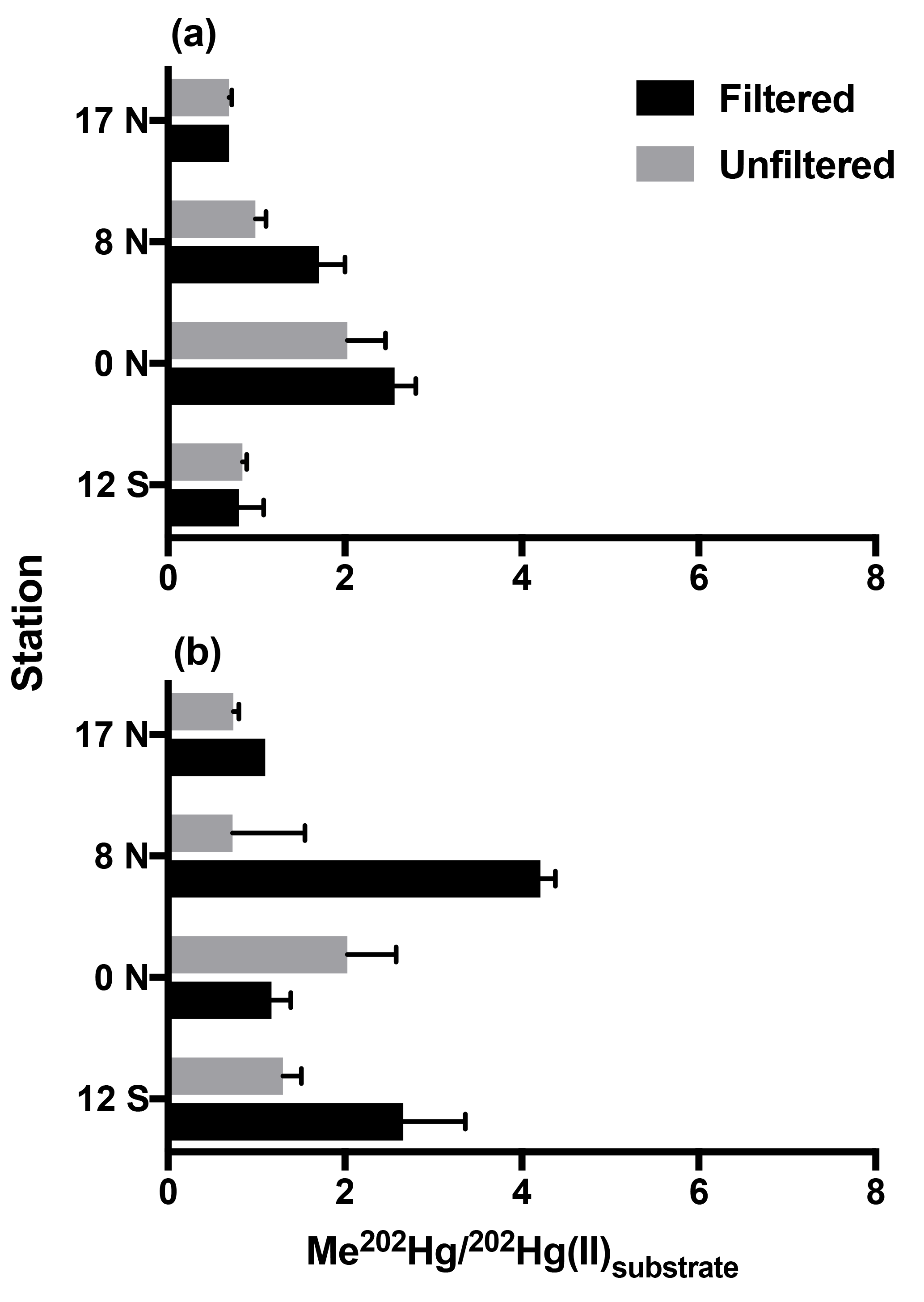
Figure 1Methylation of Hg(II) in tropical Pacific waters from triplicate bottle incubation (±1 SD), as the percentage of Me202Hg produced from excess 202Hg(II) substrate from isotope spike additions, for cruise stations from the North to South Pacific. Methylation was measured in 0.2 µm filtered and unfiltered waters from the chlorophyll-maximum (a) and oxygen-minimum (b) depths at each station.
Similar experiments in Mediterranean waters reported significant methylation in waters filtered through 0.45 µm filters, which allows the passage of some plankton and many microbes relative to unfiltered waters (Monperrus et al., 2007). Our methylation rates measured in filtered waters averaged 1.4× those of unfiltered waters (range 0.58–2.1×). This is opposite to the methylation rates measured in filtered Mediterranean waters (0.8 % d−1), which were half those in unfiltered waters (1.7 % d−1; Monperrus et al., 2007). Thus, enhanced methylation in filtered water may dominate our central Pacific station depths due to its nutrient regime while larger bacteria and perhaps eukaryotic algae may contribute in more nutrient-rich environments such as those studied by Monperrus et al. Furthermore, the relative role of noncellular versus microbial methylation may change in contrasting marine regions or due to seasonal variability. Direct comparisons cannot be made until additional rates are determined in filtered water.
In addition to enhanced methylation in filtered waters, noncellular methylation is indicated by rapid initial methylation of added 202Hg(II) spike within the elapsed time, 30–120 min, between 202Hg(II) spike addition and preservation of the t0 samples with acid (Supplement Fig. S1). This initial methylation, like that of methylation rates over 24 h incubations, was highest at 8∘ N (subsurface chlorophyll-maximum (CMX) filtered water: 1.98 % d−1), the Equator (CMX filtered water: 2.50 % d−1; oxygen-minimum (OMZ) filtered water: 1.17 % d−1), and 12∘ S (CMX unfiltered water: 1.14 % d−1), where it accounted for all methylation observed during the 24 h incubations (Fig. S1). Rapid methylation has been observed in measurements of labeled Hg(II) additions to lake water (Eckley and Hintelmann, 2006) and in previous incubations of unfiltered seawater (Lehnherr et al., 2011). Previous marine methylation rate measurements with isotopes have exclusively used unfiltered waters or filters that allow microbes to pass through (Monperrus et al., 2007; Lehnherr et al., 2011), which prevented distinction between microbial and nonmicrobial methylation in those experiments.
Table 2Potential methylation (km) and demethylation (kd) rate constants from triplicate bottle incubations of tropical Pacific waters in filtered and unfiltered water. Linear calculations were made from differences between t0 and t24 hour time points of added MM198Hg. Exponential decay was calculated from 36 h time course incubations.
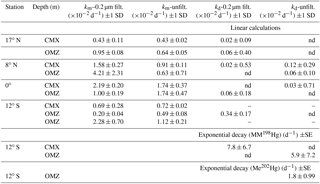
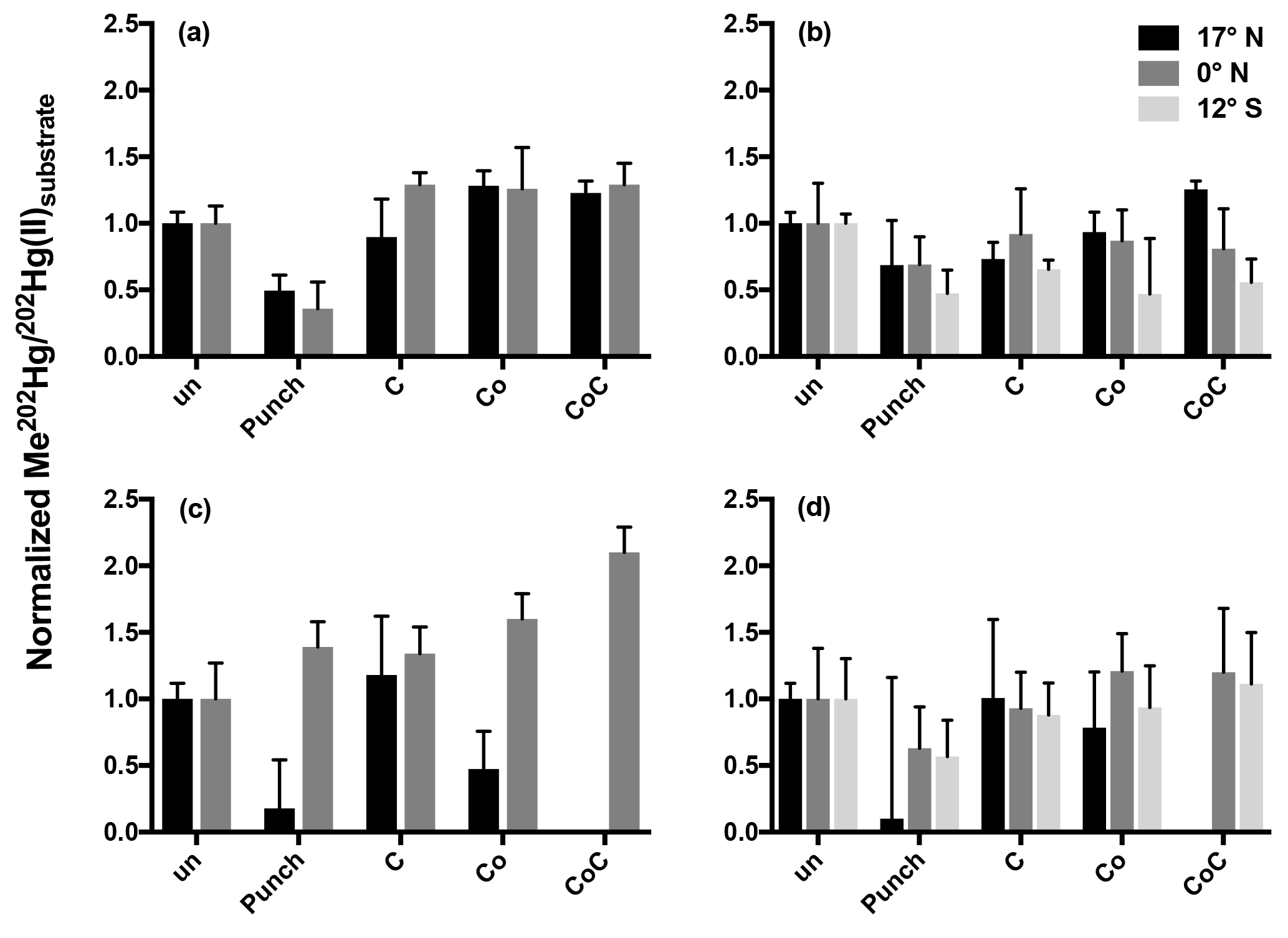
Figure 2Normalized methylation rates from 24 h incubations. Methylation rates from triplicate incubation bottles (±1 SD) normalized to unamended (un) 0.2 µm filtered (a, c) and unfiltered (b, d) water from the chlorophyll maxima (a, b) and oxygen minima (c, d). Treatments are abbreviated as follows: cobalt (Co); succinate (C); in situ pump subsamples (punch).
Methylation rates were decoupled from ambient MeHg concentrations. At chlorophyll-maximum depths, methylation rates were highest at the Equator (CMX: 2.03–2.56 % d−1) and lowest at 17 ∘ N in the North Pacific (CMX: 0.69 % d−1; Fig. 1) despite nearly identical ambient MeHg concentrations (Table 1). At oxygen-minimum depths, methylation rates also exhibit no relationship with measured MeHg concentrations, including 17∘ N in the North Pacific (OMZ: 0.74 % d−1–1.10 % d−1), where the methylation rate was lowest despite the highest MeHg concentrations (Table 1). As a result, methylation rates alone do not account for observed MeHg concentrations across gradients of productivity.
Previous dual tracer measurements have quantified simultaneous methylation and demethylation within marine waters (Monperrus et al., 2007; Lehnherr et al., 2011), which yield the persistence of MeHg species. Net methylation is indicated when MeHg concentrations predicted from steady-state methylation and demethylation rate measurements account for more than 100 % of ambient MeHg. Conversely, net demethylation is indicated when predicted MeHg concentrations account for less than 100 % of ambient MeHg. However, rapid demethylation of the added MM198Hg spike prior to t0 fixation was observed in the majority of our samples, which yielded nonquantitative demethylation rates in many cases (Table 2) due to low excess MMHg concentrations (median: 10–47 fM from all stations) remaining at measured time points despite MM198Hg spike additions in excess of 500 fM (full data in the Supplement). At the 12∘ S station, higher-resolution time point measurements yielded (Table 2) quantifiable measurements of MM198Hg, resulting in demethylation rate constants that were an order of magnitude higher (5.9 and 7.8 d−1) than those measured in Arctic waters (0.23–0.59 d−1; Lehnherr et al., 2011). These limited measurements and the demethylation that is indicated by the loss of the MM198Hg spike prior to t0 fixation suggest the importance of demethylation despite our inability to quantify demethylation rates.
3.2 Amended samples
The relative importance of methylation and demethylation in steady-state MeHg concentrations was further explored by testing whether methylation rates were limited by Hg(II) substrate supply or cofactor supply in central Pacific waters. The correlation between MeHg concentrations and rates of organic carbon remineralization in North Pacific Intermediate Water (Sunderland et al., 2009) indicates that these low-oxygen waters are poised for MeHg production from Hg(II) substrate released during remineralization. The oxygen-minimum depths targeted at each station corresponded to depths of the highest rates of organic matter remineralization (Fig. S3). However, unamended methylation rates were lowest at 17∘ N along the cruise transect despite water from this station displaying the greatest drawdown of dissolved oxygen (AOU: 283 µmol kg−1; Table 1).
We amended incubation water with filtered particulate matter, carbon (as succinate), and cobalt additions. The filtered particulate matter punches were collected from oxygen-minimum depths at each station using in situ pumps (Munson et al., 2015) to test the ability of particulate matter to increase methylation through the supply of Hg(II) substrate, thus testing whether methylation in the water column is limited by Hg(II) substrate availability. Each punch represented the particulate matter in approximately 70 L of seawater, a ∼310-fold enrichment of particles to the incubation and 0.35 to 1.05 pmol of added total Hg (Table S2). Succinate was added to distinguish between general stimulation of microbial activity and the site-specific particulate matter contained in the filter punch additions.

Figure 3Pacific water column methylated mercury (MeHg) production over 36 h at 12∘ S. Methylation rates from triplicate incubation bottles (±1 SD) in 0.2 µm filtered and unfiltered water from the chlorophyll-maximum and oxygen-minimum depths in the South Pacific. Rapid demethylation of MeHg occurred in the presence of particles in unfiltered low-oxygen water. Newly produced MeHg only persisted beyond 12 h in 0.2 µm filtered water.
Rather than relieving widespread Hg(II) limitation in low-oxygen waters, the addition of the punch material only stimulated methylation in filtered waters from the oxygen-minimum waters at the Equator over a 24 h incubation period (Fig. 2c). The addition of filtered particulate matter to unfiltered waters from other oxygen-minimum depths did not produce clear responses and high variability was seen among replicate samples (Fig. 2). Furthermore, the addition of filtered particulate matter lowered methylation rates relative to unamended samples over 24 h incubation periods. The failure to stimulate methylation with site-specific organic matter suggests that methylation at these Pacific station depths is not limited by Hg(II) substrate availability. Full Hg speciation measurements at incubation stations (Table 1; Munson et al., 2015) can be used to calculate the maximum concentration of the dissolved Hg(II) species available as methylation substrate according to Eq. (1):
Concentrations of Hg(II) calculated from Eq. (1) are relatively high (∼1 pM, Fig. S2) in low-oxygen waters at all incubation stations and comprise 41 %–90 % of the total Hg measured. These Hg(II) concentrations provide further support that methylation is not limited by Hg(II) substrate supply. Thus, neither bulk ambient Hg(II) concentrations nor increased local particulate matter supply appear sufficient as sources of Hg(II) substrate to the mechanisms involved in methylation in these waters.
Although the addition of filtered organic matter did not enhance methylation, amendments with other potential cofactors were more robust. Inorganic cobalt can limit the growth of, and MeHg production by, sulfate-reducing bacteria, including known Hg methylators (Ekstrom and Morel, 2008). Although cobalt serves as the center of methylcobalamin, the cofactor implied in hgcAB-mediated Hg methylation (Parks et al., 2013), inorganic cobalt (Co) by itself is not known to influence Hg methylation. As a result the significant increases in methylation from the combined addition of succinate and Co to oxygen-minimum waters at 12∘ S (Fig. 2) were surprising given that the greatest increase in methylation (2× that of unamended water) occurred in filtered rather than unfiltered water (Fig. 2c) where we hypothesized their addition would stimulate hgcAB-mediated methylation. The enhanced methylation in the presence of succinate and Co in the presence of minimal cellular material warrants further study as a potential abiotic mechanism of Hg methylation, such as potential competition between Co(II) and Hg(II) for organic ligand binding that could increase Hg(II) substrate availability for methylation.
3.3 Time course
Although methylation rates calculated from 24 h incubation time points (Table 2) were comparable to those measured in other marine regimes, higher temporal resolution of sampling at the South Pacific station, 12∘ S, reveals that methylation of added 202Hg(II) is rapid, with maximum methylation reached within 6 h in unfiltered waters (Fig. 3). Likewise, increased methylation in amendments with C, Co, and organic matter was rapid (Supplement Table S2). Subsequent demethylation of newly produced Me202Hg occurs within 12 h of total incubation time in incubations of unfiltered water (Fig. 3). Demethylation appeared to be a dynamic process, with the demethylation rate constant of newly produced Me202Hg (from added 202Hg(II)) in unfiltered waters calculated at 1.8 d−1 in unfiltered water from the oxygen-minimum depth (Fig. 3).
A transition between net methylation and net demethylation was observed within the first 12 h of incubation in both chlorophyll-maximum and oxygen-minimum waters (Fig. 3). This rapid transition observed at 12∘ S suggests that 24 h incubation time points are not sufficiently resolved to document these reactions in detail at our level of spike addition in central Pacific oligotrophic waters. This implies that our calculated rates (Table 2) underestimate maximum rates of methylation and demethylation that occur in situ.
Demethylation in oxygen-minimum waters from 12∘ S over the entire 36 h incubation period was influenced by the presence of particles, as shown by the demethylation of newly produced Me202Hg (Fig. 3). In contrast, methylation in filtered waters is similar in magnitude to that in unfiltered waters but persists over the course of the incubation (Fig. 3). The use of dual tracers in this time course experiment reveals that demethylation controls MeHg concentrations as the incubation approaches steady state, indicating an important role of demethylation in determining MeHg concentrations in the marine water column and its availability for bioaccumulation.
The rapid transition between MeHg production and demethylation that was observed in water from 12∘ S obscures the conclusions that can be made from 24 h incubation results (Fig. 1). Although amendment of incubated waters appeared to decrease methylation in comparison to unamended waters (Fig. 2), rapid demethylation of newly produced Me202Hg within the 24 h incubation period could produce similar results. Notably, the low MeHg production that was indicated after 24 h in unfiltered water (Fig. 2b, d) and filtered water (Fig. 2a, c) amended with filtered particulate matter could represent rapid methylation followed by demethylation kinetics similar to those observed at 12∘ S.
3.4 Mercury methylation and organic matter remineralization
Although elevated ambient MeHg concentrations have long been recognized as occurring at depths of high oxygen utilization (Mason and Fitzgerald, 1993; Cossa et al., 2009; Sunderland et al; 2009; Cossa et al., 2011; Bowman et al., 2015, 2016), gaps persist in our understanding of how organic matter remineralization influences methylation. While low MeHg concentrations in surface and deep waters are maintained due to demethylation by both photodemethylation and biological demethylation (Cossa et al., 2009; Sunderland et al., 2009; Monperrus et al., 2007; Lehnherr et al., 2011), subsurface peaks in MeHg concentrations are thought to be controlled by two factors: (1) the release of Hg(II) substrate from sinking organic matter remineralization (Cossa et al., 2009; Sunderland et al., 2009) and (2) enhancement of microbial loop activity (Heimbürger et al., 2010). Microbial loop activity has been suggested to dominate subsurface MeHg dynamics especially in oligotrophic waters (Heimbürger et al., 2010).
The subtropical gyre waters at 17 and 8∘ N were oligotrophic regions exhibiting nitrogen stress while 12∘ S was within a region exhibiting both low nitrate concentrations and iron stress (Saito et al., 2014). Concentrations of THg in the North Pacific reveal a sharp transition between North Pacific Intermediate Water and intermediate waters south of 17∘ N (Sunderland et al., 2009; Munson et al., 2015). Within the equatorial Pacific we observed an area of low Hg species concentrations. We also observed a significant reduction of Hg(II) centered at 12∘ N and extending toward the 8∘ N incubation station that corresponds to regions of denitrification between 200 and 400 m, as indicated by calculated N* values (Munson et al., 2015). The reduction of Hg(II) appears to limit the supply of Hg(II) to depths below 400 m, consistent with conceptual models of methylation due to substrate supply (Sunderland et al., 2009; Lehnherr et al., 2011).
However, the addition of filtered particulate matter from in situ pump filter punches to bottle incubation did not enhance potential methylation in waters collected from this site. These observations suggest that delivery of Hg(II) by bulk organic matter does not limit MeHg production in North Pacific waters. This observation is consistent with some field data, in which ratios of MeHg to Hg below the photic zone are not elevated compared to other regions of the water column (Sunderland et al., 2009; Hammerschmidt and Bowman, 2012), including regions adjacent to our incubation stations (Munson et al., 2015). In contrast, the addition of Co and additive additions of Co and succinate were able to promote methylation, most notably in South Pacific waters, which suggests a more specific control, such as Hg ligand binding or cofactor availability, on methylation rather than bulk Hg(II) supply. Therefore, the dominance and rapidity of initial methylation in our incubations may indicate the importance of ligand exchange with Hg(II) substrate in oligotrophic waters.
In addition to the potential role of organic matter in controlling Hg methylation, our results indicate an important role of particulate matter in demethylation and thus water column MeHg concentrations. The enriched Hg species spikes used as tracers were pre-equilibrated in filtered seawater for 24 h prior to addition to bottle incubations in order to allow Hg to bind with dissolved organic matter complexes in an attempt to more closely mimic seawater speciation of Hg (Lamborg et al., 2004). Given that the MM198Hg-enriched stock solution was stable over the 24 h pre-equilibration period in filtered seawater, the rapid demethylation observed upon addition of the spike to seawater at tracer levels in incubations was surprising. As the filtered seawater used for pre-equilibration differed from incubation water only in its ratio of enriched Hg to ligands, the rapid demethylation may further indicate the role of ligands in controlling Hg species dynamics.
We measured methylation of added 202Hg(II) substrate in filtered waters, which suggests a potential role for noncellular methylation in oligotrophic marine systems. Enhanced Hg methylation in the central Pacific Ocean filtered water implicates noncellular or extracellular processes as the primary mechanisms of mercury transformation in the marine water column. Determining the extent of this noncellular methylation relative to microbial methylation is important for understanding the potential for MMHg entry into the marine food web.
The potential for a noncellular pathway of Hg methylation dominating at these Pacific water depths is consistent with current genomic studies. Although functional hgcA and hgcB genes have been identified in marine regions that are influenced by continental shelf sediments, permafrost, and estuarine waters, functional hgcAB sequences have not been isolated from our incubation stations (Podar et al., 2015). The failure to identify functional hgcAB sequences from our incubation stations does not exclude the possibility that these genes are present in such low abundance as to escape detection or the ability of the uncharacterized sequences to encode methylation. However, our results suggest that regional differences in methylating cofactors, perhaps ligands and competing metals, and efficiency of particle-driven demethylation may be more important than hgcAB-mediated methylation in controlling MeHg concentrations in the oligotrophic Pacific.
Although we expected increased methylation following stimulation of heterotrophic bacteria following succinate addition, cellular methylation did not appear to dominate oligotrophic waters along our Pacific transect. Instead, the high methylation rates in our incubations of filtered water suggest a need to investigate the role of Hg–ligand binding in controlling steady-state MeHg concentrations. Methylation over 24 h incubations was enhanced by the addition of Co, potentially due to the activity of ligands that can interact with both Hg(II) and Co. Ligands may also influence demethylation, which appeared to control MeHg concentrations across wide gradients in productivity across this section of the Pacific. Rapid demethylation was observed in filtered water, despite pre-equilibration, and following particulate matter addition. Demethylation, and thus the persistence of MeHg in the water column, may be influenced by the relative concentration of MeHg to organic matter, both dissolved and particulate.
The full data set for incubations is available in the Supplement, Table S2.
The supplement related to this article is available online at: https://doi.org/10.5194/bg-15-6451-2018-supplement.
KMM, CHL, and MAS designed the research; KMM, CHL, and RMB performed the research; KMM and CHL analyzed data; KMM and CHL prepared the paper with contributions from RMB and MAS.
The authors declare that they have no conflict of interest.
This work was funded by the National Science Foundation in a Chemical
Oceanography Program Grant (OCE-1031271) awarded to Carl H. Lamborg and Mak A. Saito
and a graduate student fellowship to Kathleen M. Munson. We thank Vic Polidoro,
Trevor Young, Captain Drewry, and the scientific and ship's crew of the R/V Kilo Moana.
Edited by: Gerhard Herndl
Reviewed by: two anonymous referees
Black, F. J., Conaway, C. H., and Flegal, A. R.: Stability of dimethylmercury in seawater and its conversion to monomethyl mercury, Environ. Sci. Technol., 43, 4056–4062, https://doi.org/10.1021/es9001218, 2009.
Bowman, K. L., Hammerschmidt, C. R., Lamborg, C. L., and Swarr, G.: Mercury in the North Atlantic Ocean: the US GEOTRACES zonal and meridional sections, Deep Sea Res., 116, 251–261, https://doi.org/10.1016/j.dsr2.2014.07.004,116, 2015.
Bowman, K. L., Hammerschmidt, C. R., Lamborg, C. L., Swarr, G. J., and Agathar, A. M.: Distribution of mercury species across a zonal section of the eastern tropical South Pacific Ocean (US GEOTRACES GP16), Mar. Chem., 186, 156–166, https://doi.org/10.1016/j.marchem.2016.09.005, 2016.
Celo, V., Lean, D. R. S., and Scott, S. L.: Abiotic methylation of mercury in the aquatic environment, Sci. Total Environ., 368, 126–137, https://doi.org/10.1016/j.scitotenv.2005.09.043, 2006.
Cossa, D., Averty, B., and Pirrone, N.: The origin of methylmercury in open Mediterranean waters, Limnol. Oceanogr., 54, 837–844, https://doi.org/10.4319/lo.2009.54.3.0837, 2009.
Cossa, D., Heimbürger, L.-E., Lannuzel, D., Rintoul, S. R., Butler, E. V. C., Bowie, A. R., Averty, B., Watson, R. J., and Remenyi, T.: Mercury in the Southern Ocean, Geochim. Cosmochim. Ac., 75, 4037–4052, https://doi.org/10.1016/j.gca.2011.05.001, 2011.
Drevnick, P. E., Lamborg, C. H., and Horgan, M. J.: Increase in mercury in Pacific yellowfin tuna, Environ. Toxicol. Chem., 34, 931–934, https://doi.org/10.1002/etc.2883, 2015.
Eckley, C. S. and Hintelmann, H.: Determination of mercury methylation potentials in the water column of lakes across Canada, Sci. Total Environ., 368, 111–125, https://doi.org/10.1016/j.scitotenv.2005.09.042, 2006.
Ekstrom, E. B. and Morel, F. M. M.: Cobalt limitation of growth and mercury methylation in sulfate-reducing bacteria, Environ. Sci. Technol., 42, 93–99, https://doi.org/10.1021/es0705644, 2008.
Gilmour, C. C., Podar, M., Bullock, A. L., Graham A. M., Brown, S. D., Somenahally, A. C., Johs, A., Hurt Jr., R. A., Bailey, K. L., and Elias, D.A.: Mercury methylation by novel microorganisms from new environments, Environ. Sci. Technol., 47, 11810–11820, https://doi.org/10.1021/es403075t, 2013.
Gilmour, C. C., Bullock, A. L., McBurney, A., Podar. M., and Elias, D. A.: Robust mercury methylation across diverse methanogenic Archaea, mBio, 9, e02403-17, https://doi.org/10.1128/mBio.02403-17, 2018.
Gionfriddo, C. M., Tate, M. T., Wick, R. R., Schultz, M. B., Zemla, A., Thelen, M. P., Schofield, R., Krabbenhoft, D. P., Holt, K. E., and Moreau, J. W.: Microbial mercury methylation in Antarctic sea ice, Nat. Microbiol., 1, 16127–16138, https://doi.org/10.1038/nmicrobiol.2016.127, 2016.
Haller, C. M., Rolleke, S., Vybiral, D., Witte, A., and Velimirov, B.: Investigation of 0.2 µm filterable bacteria from the Western Mediterranean Sea using a molecular approach: dominance of potential starvation forms, FEMS Microbiol. Ecol., 31, 153–161, 2000.
Hammerschmidt, C. R. and Bowman, K. L.: Vertical methylmercury distribution in the subtropical North Pacific Ocean, Mar. Chem., 132–133, 77–82, https://doi.org/10.1016/j.marchem.2012.02.005, 2012.
Heimbürger, L.-E., Cossa, D., Marty, J. C., Migon, C., Averty, B., Dufour, A., and Ras, J.: Methyl mercury distributions in relation to the presence of nano and picophytoplankton in an oceanic water column (Ligurian Sea, North-western Mediterranean), Geochim. Cosmochim. Ac., 74, 5549–5559, https://doi.org/10.1016/j.gca.2010.06.036, 2010.
Hintelmann, H. and Ogrinc, N.: Determination of stable mercury isotopes by ICP/MS and their application in environmental studies, ACS Symp. Ser., 835, 321–338, https://doi.org/10.1021/bk-2003-0835.ch021, 2003.
Lamborg, C. H., Fitzgerald, W. F., Skoog, A., and Visscher, P. T.: The abundance and source of mercury-binding organic ligands in Long Island Sound, Mar. Chem., 90, 151–163, https://doi.org/10.1016/j.marchem.2004.03.014, 2004.
Lamborg, C. H., Hammerschmidt, C. R., Gill, G. A., Mason, R. P., and Gichuki, S: An intercomparison of procedures fro the determination of total mercury in seawater and recommendations regarding mercury speciation during GEOTRACES cruises, Limnol. Oceaogr.-Meth., 10, 90–100, https://doi.org/10.4319/lom.2012.10.90, 2012.
Lamborg, C. H., Hammerschmidt, C. R., Bowman, K. L., Swarr, G. J., Munson, K. M., Ohnemus, D. C., Lam, P. J., Heimbürger, L.-E., Rijkenberg, M. J. A., and Saito, M. A.: A global ocean inventory of anthropogenic mercury based on water column measurements, Nature, 512, 65–68, https://doi.org/10.1038/nature13563, 2014.
Lehnherr, I., St. Louis, V. L., Hintelmann, H., and Kirk, J. L.: Methylation of inorganic mercury in polar marine waters, Nat. Geosci., 4, 298–302, httpsL://doi.org/10.1038/ngeo1134, 2011.
Li, W. K. W. and Dickie, P. M.: Growth of bacteria in seawater filtered through 0.2 µm Nucleopore membranes: implications for dilution experiments, Mar. Ecol.-Prog. Ser., 26, 245–252, 1985.
Mason, R. P. and Fitzgerald, W. F.: The distribution and cycling of mercury in the equatorial Pacific Ocean, Deep-Sea Res. Pt. I, 40, 1897–1924, https://doi.org/10.1016/0967-0637(93)90037-4, 1993.
Monperrus, M., Tessier, E., Amouroux, D., Leynaert, A., Huonnic, P., and Donard, O. F. X.: Mercury methylation, demethylation and reduction rates in coastal and marine surface waters of the Mediterranean Sea, Mar. Chem., 107, 49–63, https://doi.org/10.1016/j.marchem.2007.01.018, 2007.
Munson, K. M., Babi, D., and Lamborg, C. H.: Monomethylmercury determination from seawater using ascorbic-acid assisted direct ethylation, Limnol. Oceanogr.-Meth., 12, 1–9, https://doi.org/10.4319/lom.2014.12.1, 2014.
Munson, K. M., Lamborg, C. H., Swarr, G. J., and Saito, M. A.: Mercury species concentrations and fluxes in the Central Tropical Pacific Ocean, Global Biogeochem. Cy., 29, 656–676, https://doi.org/10.1002/2015GB005120, 2015.
Parks, J. M., Johs, A., Podar, M., Bridou, R., Hurt Jr, R. A., Smith, S. D., Tomanicek, S. J., Qian, Y., Brown, S. D., Brandt, C. C., Palumbo, A. V., Smith, J. C., Wall, J. D., Elias, D. A., and Liang, L.: The genetic basis for bacterial mercury methylation, Science, 339, 1332–1335, https://doi.org/10.1126/science.1230667, 2013.
Podar, M., Gilmour, C. C., Brandt, C. C., Soren, A., Brown, S. D., Crable, B. R., Palumbo, A. V., Somenahally, A. C., and Elias, D. A.: Global prevalence and distribution of genes and microorganisms involved in mercury methylation, Sci. Adv., 1, e1500675, https://doi.org/10.1126/sciadv.1500675, 2015.
Rappé, M. S., Connon, S. A., Vergin, K. L., and Giovannoni, S. J.: Cultivation of the ubiquitous SAR11 marine bacterioplankton clade, Nature, 418, 630–633, https://doi.org/10.1038/nature00917, 2002.
Saito, M. A., McIlvin, M. R., Moran, D. M., Goepfert, T. J., DiTullio, G. R., Post, A. F., and Lamborg, C. H.: Multiple nutrient stresses at intersecting Pacific Ocean biomes detected by protein biomarkers, Science, 345, 1173–1177, https://doi.org/10.1126/science.1256450, 2014.
Sunderland, E. M., Krabbenhoft, D. P., Moreau, J. W., Strode, S. A., and Landing, W. M.: Mercury sources, distribution, and bioavailability in the North Pacific Ocean: insights from data and models, Global Biogeochem. Cy., 23, 1–14, https://doi.org/10.1029/2008GB003425, 2009.
Yin, Y., Li, Y., Tai, C., and Jian, G.: Fumigant methyl iodide can methylate inorganic mercury species in natural waters, Nat. Commun., 5, 4633, https://doi.org/10.1038/ncomms5633, 2014.