the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Jul 2026
| 14 Jul 2026
Technical note: Kinetically resolved volatile and redox fingerprints of geologic materials by ramped combustion microchromatography
Shuzhuang Wu
Samuel L. Jaccard
Matthieu E. Galvez
The biogeochemical cycles of carbon, oxygen and sulfur are fundamentally interlinked, yet quantifying the reactivity of these elements within complex geological matrices remains a major analytical challenge. We present a novel integrated TGA/DSC-MicroGC system that simultaneously monitors mass loss, heat flow, and evolved gas composition during controlled heating in a gas mixing furnace. This approach kinetically resolves and quantifies distinct carbon and sulfur materials through their thermal decomposition profiles. Furthermore, continuous monitoring of oxygen consumption provides a direct measure of a material's oxidability in various temperature windows, a redox fingerprint. Validation against geochemical standards and application to sediments from the Congo Basin and Alpine Lake Cadagno (Switzerland) reveal diagenetic transitions and paleoenvironmental fluxes that are invisible to conventional bulk methods. This integrated methodology provides a mechanistic, high-resolution insight into electron-transfer processes in natural materials. This offers new avenues for probing biogeochemical cycling, redox evolution and environmental reactivity across Earth systems.
- Article
(4123 KB) - Full-text XML
-
Supplement
(481 KB) - BibTeX
- EndNote




The fate of carbon (C) and sulfur (S) in sediments depends not only on their bulk concentration, but also on their molecular speciation and intrinsic reactivity (Alt and Shanks III, 2006; Galvez, 2020; Hayes and Waldbauer, 2006; Paytan et al., 1998). For example, the oxidation of labile biomolecules occurs over temperature windows distinct from that of refractory C phases, such as graphite. Similarly, monosulfides display oxidation profiles that differ from those of crystalline sulfides such as pyrite (Boudreau, 1992; Hemingway et al., 2018; Ordoñez et al., 2019; Sebag et al., 2016). Their combustion enthalpy and O2 demand differ too, reflecting the specific bonding environment and redox state of each compound or mineral phase (Galvez and Jaccard, 2021). Existing analytical techniques are poorly suited to capture these key dimensions of reactivity. Standard methods, such as elemental analysis or X-ray fluorescence, quantify total C and S, but are blind to molecular speciation and oxidation behavior (Berg et al., 2022; Carter et al., 2024; Salonen, 1979; Yoon et al., 2018; Zhao et al., 2020). Meanwhile, Rock-Eval, a widely used tool in organic petrology, provides bulk chemical and kinetic parameters for hydrocarbon source rocks (Espitalie et al., 1985). However, its focus is primarily on the ramped pyrolysis and oxidation of carbon-bearing materials, and only the latest generation, Rock-Eval 7, allows sulfur to be characterized simultaneously (Cohen-Sadon et al., 2022). Moreover, Rock-Eval does not directly link thermal degradation and combustion profiles to their underlying oxygen demands, which is a key parameter for quantifying the redox state and oxidation potential of rocks and sediments.
Recent advances, such as the high-temperature titration method of Galvez and Jaccard (2021), have successfully quantified the total redox capacity of natural samples, partially bridging the gap between compositional and redox characterization. But this protocol was designed as an endpoint measurement and, as such, does not kinetically resolve the oxidation process, which we define here as the “oxidability” of the sample: the temperature-dependent distribution of oxygen consumption, mass loss heat release and gas production during controlled heating In addition, the endpoint approach does not provide direct insight into the individual contributions of C, S, and redox-active iron (Fe) species – including iron locked in mineral lattices (e.g., Fe(II) in silicates, oxides, and sulfides such as pyrite), as well as iron complexed within or adsorbed onto organic biomass – to the overall oxygen demand (Galvez and Jaccard, 2021; Galvez et al., 2025). Consequently, a major disconnect persists between our understanding of global redox cycles and our ability to characterize the combustion kinetics of the materials that drive them. To address this gap, an analytical approach is required that goes beyond bulk characterization, providing a simultaneous, kinetically resolved view of mass loss, heat flow, oxygen consumption and gas evolution during ramped pyrolysis or combustion.
We have developed such a platform: a novel integrated system combining thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and micro gas chromatography (MicroGC). This configuration enables real-time, correlated measurements of mass loss, heat flow, and the evolving composition of gases, including O2, CO2, SO2, H2S, COS, throughout controlled thermal decomposition and oxidation. Specifically, this integration enables two key innovations:
-
It distinguishes thermally reactive C and S pools based on their characteristic decomposition or oxidation profiles and activation energies.
-
It measures oxygen demand during combustion in a continuous, kinetically resolved set-up by directly monitoring oxygen consumption, yielding a unique chemical, thermal and redox fingerprint for each sample.
Together, these capabilities reveal a new means of characterizing redox reactivity. By linking mass loss, heat flow, gas evolution and oxygen demand, this approach can reveal diagenetic pathways and paleoenvironmental signatures in complex natural archives that remain inaccessible to conventional bulk analytical techniques.
2.1 Sample selection and preparation
Sediment samples for TGA/DSC-MicroGC were obtained from archived freeze-dried material. We have analyzed a range of representative sediment materials containing C, S and hydrogen (H)-bearing compounds from contrasting depositional environments: the Congo Basin, Democratic Republic of Congo (DRC) and Lake Cadagno, Switzerland. The Congo Basin hosts the world's largest tropical peatland complex (Crezee et al., 2022; Garcin et al., 2022). These sediments provide a natural archive for studying organic matter degradation pathways associated with peat burial and climate-driven shifts since the Last Glacial Maximum. In contrast, Lake Cadagno, a meromictic lake in Switzerland with a permanently anoxic deep layer, is an ideal natural laboratory for studying microbial redox processes and their long-term preservation in the sedimentary record (Berg et al., 2022; Berg et al., 2025; Janssen et al., 2022; Dupeyron et al., 2025). Our approach illuminates the evolving speciation and reactivity of C-and S-bearing phases across distinct sedimentary redox environments.
2.2 Instrumentation and ultra-fast configuration mode
We coupled a TGA/DSC 3+ (Mettler Toledo) with a MicroGC 990 (Agilent SRA Instruments) (Fig. 1) and calibrated the integrated system for precise, semi-continuous H-, C-, O- and S-bearing gases (Fig. 3). The TGA/DSC simultaneously monitors mass and heat flow changes as a function of temperature within a programmable continuous-flow gas-mixing furnace. The MicroGC, a gas chromatograph equipped with short columns, separates, identifies, and quantifies light volatile compounds, including H2O, O2, CH4, CO2, H2S, COS and SO2, present in the evolving gas mixture. In our hybrid setup, samples are heated in a programmable furnace at 10 °C min−1 from 100–1000 °C under a controlled atmosphere (typically 10 mL min−1 N2 and 1 mL min−1 O2). A secondary oxidation furnace downstream of the primary furnace, maintained at temperatures between 300–800 °C was used to promote complete oxidation of evolved volatile species across the heating ramp. All carrier gases (N2, O2 and He) are of ultra-high purity (>5.8; Carbagas and Linde).
Evolved gases are transferred via a heated transfer line (80 °C) to the MicroGC, equipped with Molsieve 5 Å (for N2, O2) and PORAPLOT PPU (for CO2, H2S and SO2) columns for efficient separation and quantification. The integrated TGA/DSC-MicroGC analytical system therefore provides simultaneous, temperature-resolved measurements of mass loss, heat flow, and gas composition enabling real-time characterization of oxidation and redox processes.
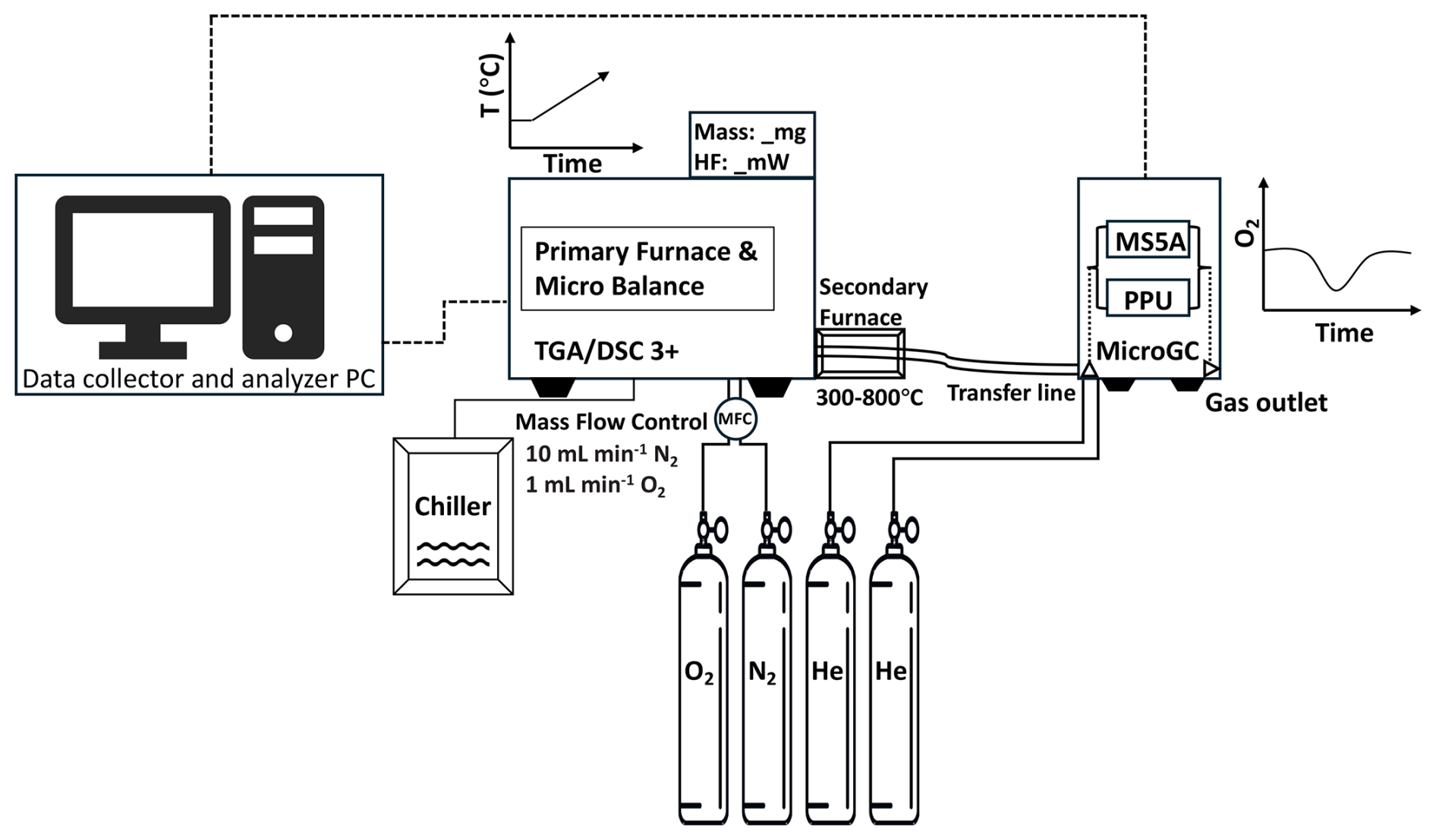
2.3 Ultra-fast chromatographic mode
To resolve discrete devolatilization events, we configured our Agilent Micro GC990 Solia system for ultra-fast dual-channel operation with a total cycle time of 54 s, comprising 15 s of sampling time and 39 s of chromatographic separation. The objective was not to maximize separation of light or heavy hydrocarbons, as is typically prioritized in chromatographic cycles lasting more than two minutes, but rather to maximize sampling density and capture transient devolatilization profiles of light molecules at high temporal resolution. The following ultra-fast MicroGC configuration prioritized cycle speed while maintaining robust peak separation for the target gases.
Channel A employed an MS5A SS column (10 m ×0.25 mm) held at 100 °C and 2.30 bar He pressure, with the injector heated at 90 °C. A 15 ms injection time was used for sharp sample introduction and backflush was initiated after 13 s of chromatography to purge retained species. This setup optimized rapid separation and detection of permanent gases (O2, N2, CO) using a thermal conductivity detector (TCD). Channel B used a PORAPLOT U FS column (10 m ×0.32 mm) maintained at 98 °C and 2.30 bar column pressure, with the injector heated at 90 °C. A 50 ms injection to ensure adequate signal to noise ratio for volatile species at low concentrations, and backflush was initiated after 10 s. This configuration enabled fast elution and quantification of CO2, H2S, H2O, and SO2.
This configuration, refined empirically through repeated optimization, delivers a complete gas composition datapoint every 54 s. A typical 90 min experiment therefore generates approximately 100 high-density gas-evolution spectra, sufficient to capture transient volatile release profiles with high temporal resolution.
2.4 Data processing and kinetic parameter estimation
Raw thermogravimetric and calorimetric data were initially processed using Mettler Toledo STARe software. Prior to kinetic fitting, baseline drift and buoyancy effects were corrected by subtracting blank runs performed under identical analytical conditions. We quantified evolved gas concentrations by external calibration against known standards and processed the MicroGC chromatograms with the Solia software (Fig. 3).
Bulk mass-loss measurement alone does not capture the intrinsic reactivity of complex materials. To overcome this limitation, our approach uses pyrograms, heat-flow data and gas-evolution profiles to characterize the reaction kinetics of C and S devolatilization and/or combustion pathways based on Arrhenius-type behavior (Eq. 1).
where k is the temperature-dependent rate constant, A is the empirically derived Arrhenius pre-exponential (“frequency”) factor, Ea is the activation energy, R is the ideal gas constant, and T is the measured temperature (see Table 1 for symbol descriptions).
Table 1List of mathematical symbols used throughout this study.

To extract kinetic parameters, we employed a dual-model approach:
-
Model-Free Isoconversional Method: First, apparent activation energies (aEa) were determined using the model-free isoconversional method of Vyazovkin (Vyazovkin and Wight, 1999), implemented within the kinetics evaluation module of the STARe software. This reaction was evaluated the across discrete fractional conversion intervals (α). This approach does not require prior assumptions about the reaction model and served as our baseline for parameter validation.
-
Distributed Activation Energy Model (DAEM): Second, we compared the isoconversional results with those obtained using a parallel-reaction Distributed Activation Energy Model (DAEM), following the methodology outlined by Hemingway et al. (2017). A critical step in our parameterization was the treatment of the pre-exponential factor (A). Use of a fixed A value led to systematic overestimation of activation energies for certain standards such as calcite that do not follow a first-order decomposition kinetics (Fig. 4). Consequently, A was iteratively optimized across a boundary range of 105 to 1012 s−1 to ensure consistency between DAEM-derived apparent activation energies to those obtained using the isoconversional method. This optimized protocol was subsequently applied to all natural samples.
3.1 Method validation: thermal signatures and calibration
The integrated analytical system produces distinct thermal decomposition and combustion signatures for a range of C- and S-bearing standards, allowing robust characterization of their thermal reactivity and associated gas-evolution patterns (Fig. 2). Organic standards, such as L-cystine and cellulose, exhibited multi-stage decomposition profiles (Fig. 2A, B). L-cystine, a sulfur-rich amino acid, decomposed primarily between 200 and 300 °C under N2 / O2 (10 : 1 vol : vol) mix atmosphere, releasing CO2, H2S, and COS (Fig. 2A, H). Cellulose combusted between 250 and 350 °C, releasing primarily CO2 and H2O (Fig. 2B, I). The IFPEN-160 000 (IFP) standard displays overlapping mass-loss events, reflecting the concurrent oxidation of organics and devolatilization of carbonates (a non-redox decarbonation reaction) (Fig. 2C, J). In contrast, inorganic standards yielded sharper and more distinct thermal spectra. Calcium carbonate (CaCO3), for instance, exhibited a single, well-defined mass loss of ∼44 % between 600 and 800 °C, consistent with its thermal decomposition into CaO and CO2 (Fig. 2D, K).
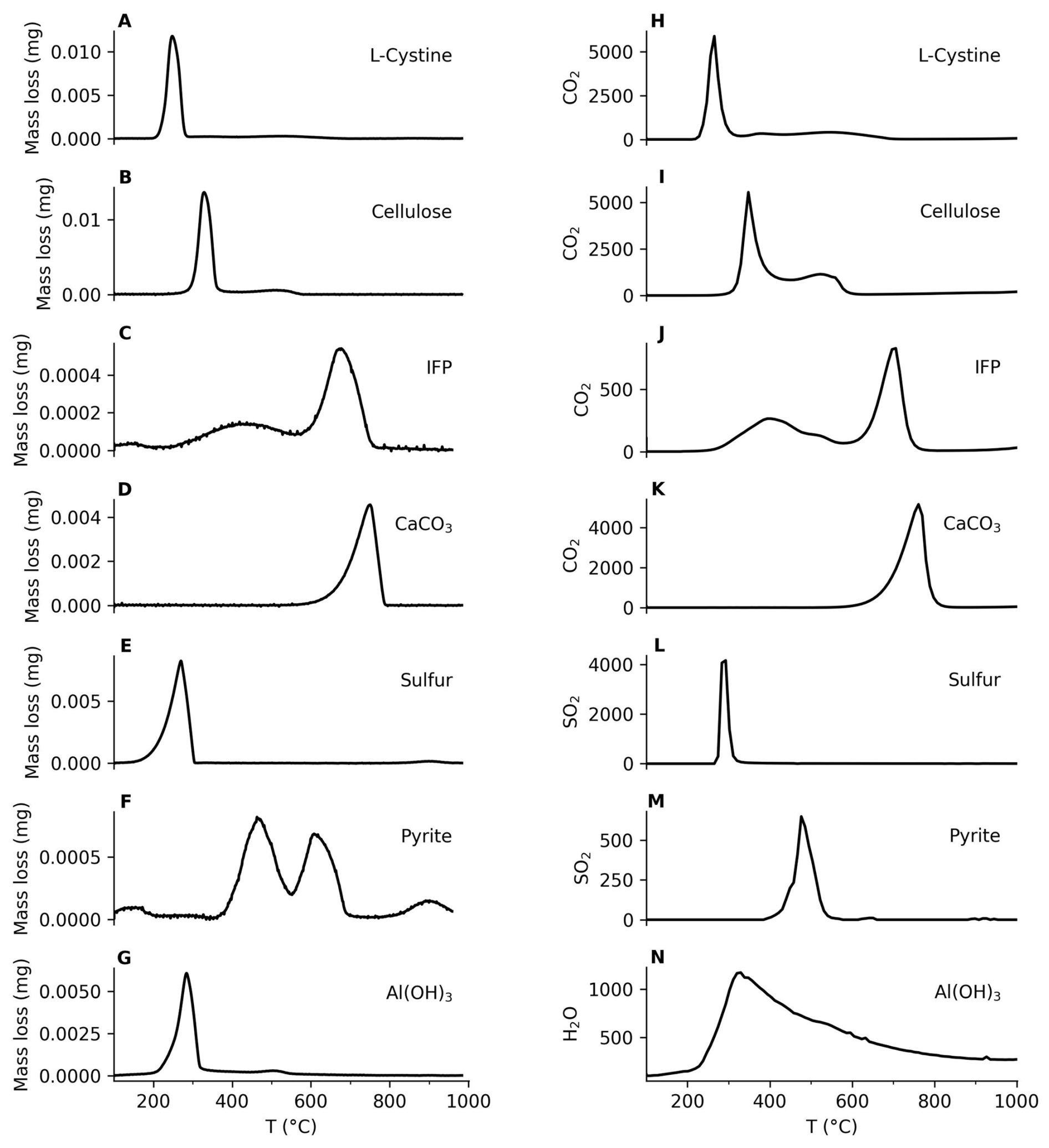
Figure 2Mass loss and devolatilized components from standards evaluated by using TGA/DSC-MicroGC system. (A, H) L-Cystine; (B, I) Cellulose; (C, J) IFP; (D, K) CaCO3; (E, L) Elemental sulfur; (F, M) Pyrite; (G, N) Al(OH)3.
Elemental S underwent rapid volatilization followed by partial oxidation to SO2 between 100 and 300 °C under oxidative condition (Fig. 2E, L). In contrast, pure pyrite (FeS2) showed a distinct oxidative mass loss between 400 and 600 °C, corresponding to its conversion to Fe2O3 and release of SO2 (Fig. 2F, M). Aluminium hydroxide [Al(OH)3] released structural water (∼13 % mass loss) between 200 and 300 °C as it dehydrated to Al2O3 (Fig. 2G, N). The combined TGA/DSC-MicroGC profiles thus differentiate organic from inorganic C- and S-bearing species based on their unique thermal decomposition kinetics or oxidation behaviour and diagnostic gas-phase reaction products. This kinetically resolved information is not accessible with conventional bulk characterization techniques.
We operated the hybrid system in oxidation mode, enabling accurate quantitative determinations of H-, C- and S-bearing volatile production and thereby allowing estimation of the corresponding reactive H, C and S contents. Under these conditions, reactive H-, C-, S-bearing species are completely oxidized to H2O, CO2 and SO2, respectively, allowing direct quantification from gas-evolution signals. H2O was calibrated by dehydrating Al(OH)3 under controlled conditions (Fig. 3A). CO2 calibration was performed using multiple certified reference materials to ensure accuracy and linearity across diverse matrices and CO2 generation mechanisms. Synthetic vitreous carbon, natural graphite, and the IFP standard were combusted in oxidation mode; pure CaCO3 was thermally decomposed to release structural carbonate CO2; and ambient air (427 ppm CO2) served as an independent gas-phase standard (Fig. 3B). Sulfur species were calibrated using the oxidation of pyrite, IFP, CLB_1 and JSD_1 standards, yielding well defined SO2 peaks (Fig. 3C). The resulting calibration curves for H2O, CO2 and SO2 exhibit excellent linearity (R2>0.99), confirming the robustness and precision of quantitative measurements in oxidation mode (Fig. 3).
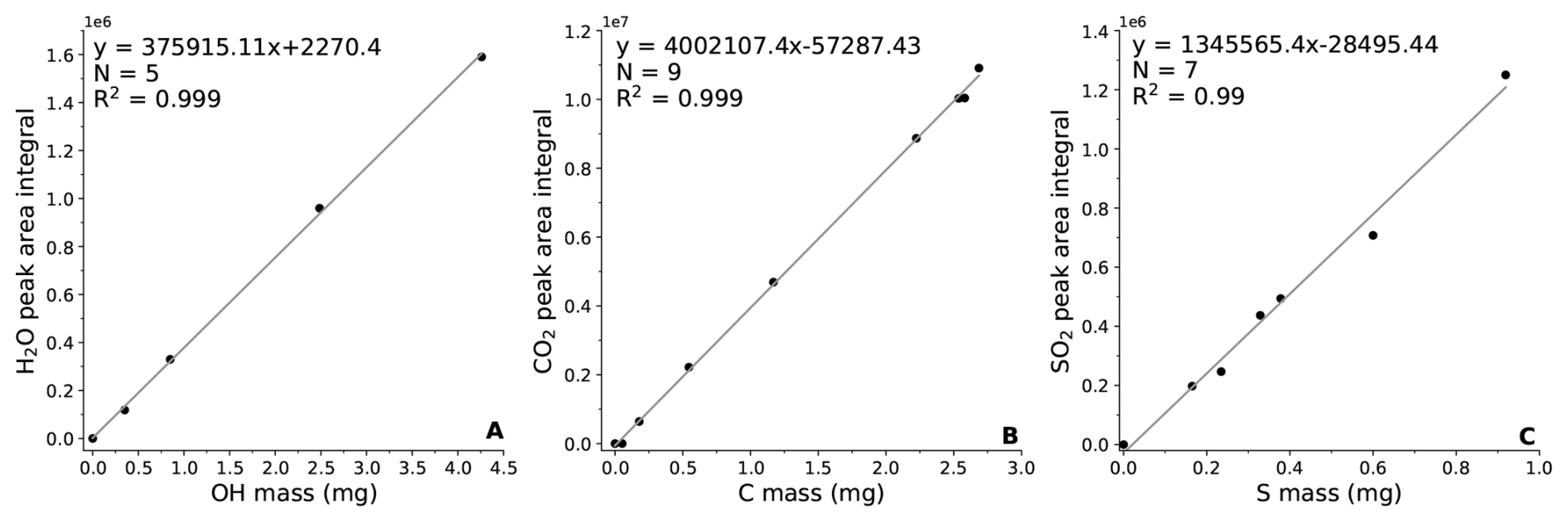
Figure 3Gas concentration calibrations. (A), Hydrogen; (B), Carbon; (C), Sulfur. Note: The different mass ranges for H, C and S calibration due to our calibrations focus on the typical ranges of H, C, S concentrations in the natural samples. S is typically low, while H and C can reach much higher concentrations in rocks and sediments, so the calibration reflects this wider range (Table S2 in the Supplement). The different slopes are reflecting different behaviors of the elements and response of the sensor.
3.2 Kinetically-resolved characterization of carbon and sulfur in natural samples
To test the kinetic approach, we first analyzed CaCO3 (99.9 % purity) using the model-free isoconversional method (Fig. 4A–D). The method yielded a reaction enthalpy (ΔH) of 177 kJ mol−1 and aEa of 164 kJ mol−1, both consistent with published values (L'vov, 1997). In contrast, the DAEM with a fixed pre-exponential factor (A=1010 s−1) produced an overestimated aEa of ∼250 kJ mol−1. Adjusting A to 105.5 s−1 resulted in a aEa of 162 kJ mol−1, closely matching the isoconversional estimate and literature values (Fig. 4D). Applying this approach to a natural sediment from the Congo Basin (Fig. 4E–H) revealed two distinct modes with aEa=160 and 120 kJ mol−1, respectively. These modes are consistent with a multi-step decomposition process involving organic matter fraxtions with contrasting thermal reactivity. The DAEM with fixed A=1010 s−1 again overestimated activation energies (∼180 and 140 kJ mol−1), whereas an optimized prefactor of A=108.5 s−1 produced values comparable to the isoconversional results. These results demonstrate that the pre-exponential factor is not a universal constant across mineral and sedimentary matrices, but depends on sample structure, crystallinity, reaction pathway and the nature of the reacting phase. Careful parametrization is therefore required, particularly for natural samples containing mixed organic and mineral phases.
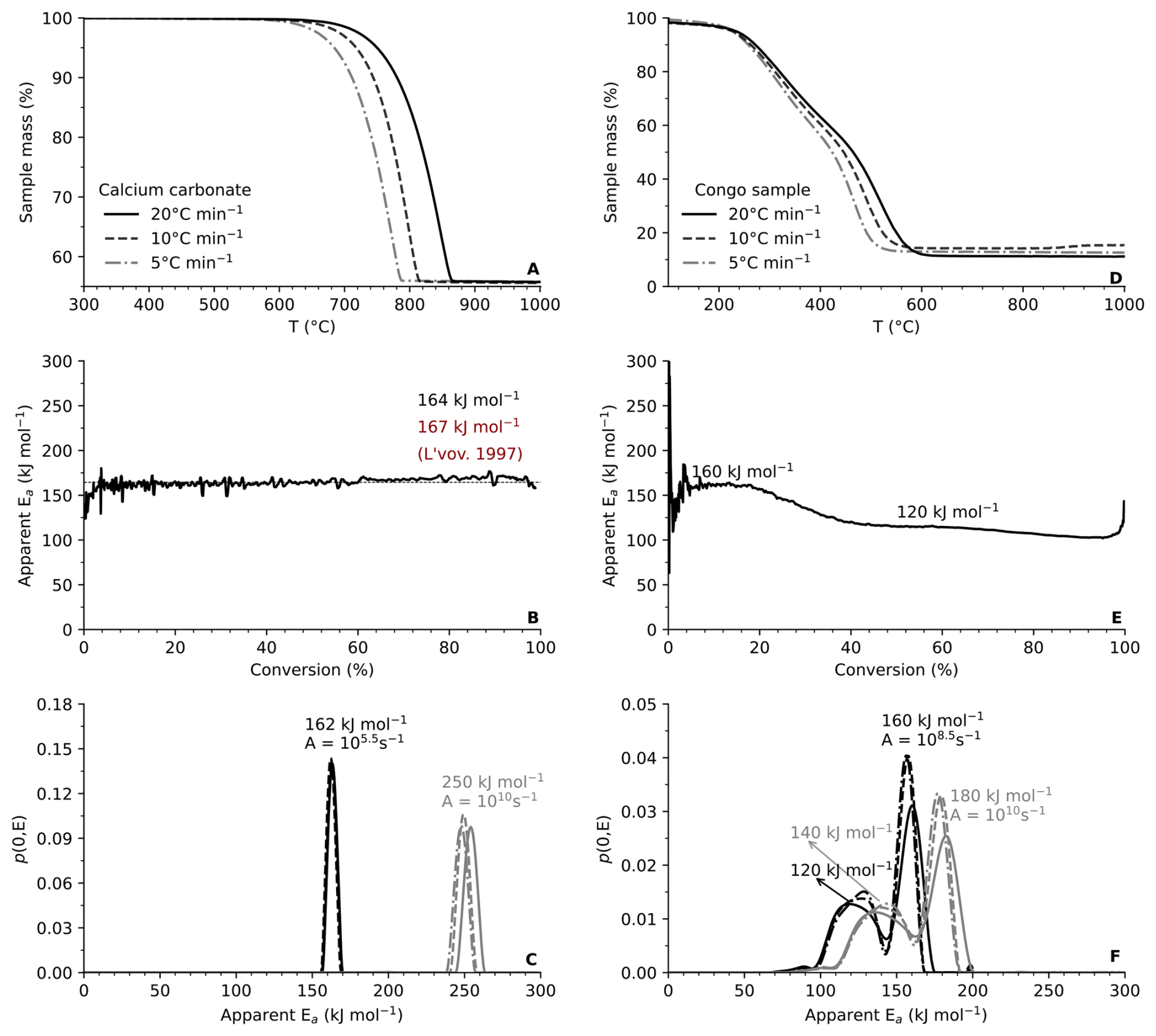
Figure 4Thermal properties for standard calcium carbonate (left) and natural sample from Congo basin (right). (A, D) Mass loss curves from thermogravimetric analysis (TGA) at heating rates of 5, 10, and 20 °C min−1; (B, E) Apparent activation energy (aEa) as a function of conversion determined via the model-free isoconversional method (Vyazovkin and Wight, 1999); (C, F) We derive the apparent activation energy distribution p(0,E) from the distributed activation energy model (DAEM) for various pre-exponential factors (A) following (Hemingway et al., 2017).
Thermograms of peatland sediment from the Congo Basin reveal two predominant organic fractions: a thermally labile component (L) with a decomposition peak near 370 °C and an aEa of ∼120 kJ mol−1, and a more refractory fraction (R), consistent with more aromatic, thermally stable organic matter, peaking at 500 °C with an aEa of ∼160 kJ mol−1 (Fig. 5A). The ratio of the labile-to-refractory peak areas (L R) increases markedly following peat initiation, indicating a higher proportion of thermally labile organic material. This substantial increase suggests either a shift in organic matter sources and/or enhanced preservation of labile compounds within the peat profile. This interpretation would be consistent with independent paleoenvironmental proxies that point to a transition to wetter conditions that promoted peat accumulation and selective preservation of labile organic matter (Garcin et al., 2022).
Similarly, thermograms from Lake Cadagno sediments reveal three distinct S-bearing fractions. Hydrogen sulfide (H2S) and carbonyl sulfide (COS) evolved at relatively low temperatures, with aEa values of approximately 110 and 130 kJ mol−1, respectively. This behaviour is consistent with the decomposition of labile organic sulfur compounds, such as thiols, sulfides, and disulfides associated with fresh organic matter (Fig. 5B). A higher temperature SO2 peak with an aEa value of approximately 150 kJ mol−1 reflects the oxidative decomposition of pyrite. This differentiation clearly separates low-temperature organic S-bearing pools from more thermally stable inorganic sulfide minerals.
The low-temperature release of H2S and COS reflects partial sulfurization associated with dissimilatory sulfate reduction (DSR), the dominant anaerobic microbial pathway for S cycling in the lake's euxinic bottom waters. Conversely, the higher-temperature SO2 peak reflects the thermal stability of pyrite, the end product of long-term biological sulphate reduction and diagenetic mineralization. Together, these results demonstrate that the TGA/DSC-MicroGC approach yields quantitative, kinetically resolved signatures of microbial sulfur cycling and organic matter transformation, linking molecular-scale reactivity to environmental processes.
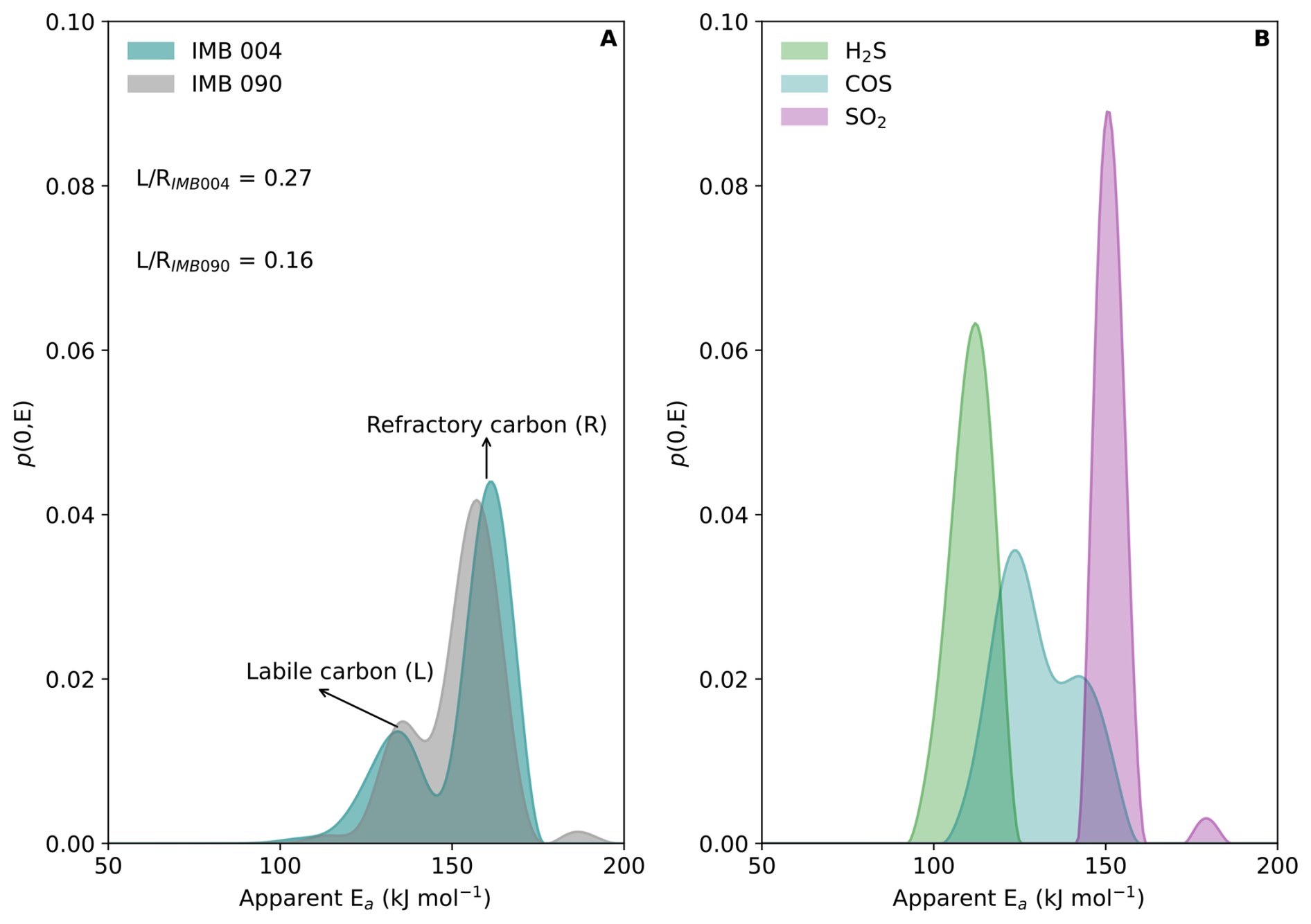
Figure 5Kinetically-resolved carbon and sulfur speciation. (A), aEa distributions for carbon speciation in peat samples from the Congo Basin, including labile and refractory carbon fractions. (B), aEa distributions of sulfur speciation in a sample from Lake Cadagno, including H2S, COS, and SO2.
3.3 Absolute Redox Capacity and Oxidation Profiles
Having established the capacity to kinetically resolve individual species, we next address a more integrated property: the specific redox capacity, dO2. To move beyond thermodynamic proxies and bulk speciation, we developed a method to quantify the oxygen demand during combustion, through time-resolved monitoring of oxygen consumption during a controlled heating ramp. Previous work has determined the redox capacity by measuring the total oxygen exchanged between a solid-state oxygen donor and the sample at elevated T under vacuum (Galvez and Jaccard, 2021; Galvez et al., 2020). But this approach is limited to endpoint measurements. Our method overcomes this limitation by continuously measuring O2 concentrations at high temporal resolution during the heating ramp. Samples are heated at a controlled rate (e.g. 10 °C min−1), under a specific gas mixture, inducing progressive devolatilization and oxidation while the MicroGC continuously monitors the evolving gas composition. The decline in the O2 concentrations is integrated over time to calculate the cumulative volume of O2 consumed, which is then normalized to the initial sample mass and the evaluated relative to the stoichiometric oxidation reactions of the major gas-producing components (Fig. 6A). This approach yields an absolute oxygen demand while simultaneously preserving the temperature-resolved oxidation profile. It therefore provides both the total redox capacity and the distribution of that capacity across distinct thermal domains. The method shows high reproductivity with a relative standard deviation of 0.82 % for vitreous carbon standards. Importantly, our results are consistent with those obtained by the vacuum line protocol (Galvez and Jaccard, 2021), validating the precision and robustness of this continuous, kinetically resolved redox quantification method.
Redox capacity (dO2) was calculated as:
- i.
Volume of O2 supply (Vsup)
where f is the O2 flow rate (1 ml min−1) and Δt is time interval.
- ii.
Volume of O2 consumed (Vcon)
where Aeff is time-integrated effective area corresponding to consumed O2 and Atot is time-integrated total area.
- iii.
Redox capacity (dO2)
where P is gas pressure, R is the ideal gas constant, T is the room temperature and m is sample mass.
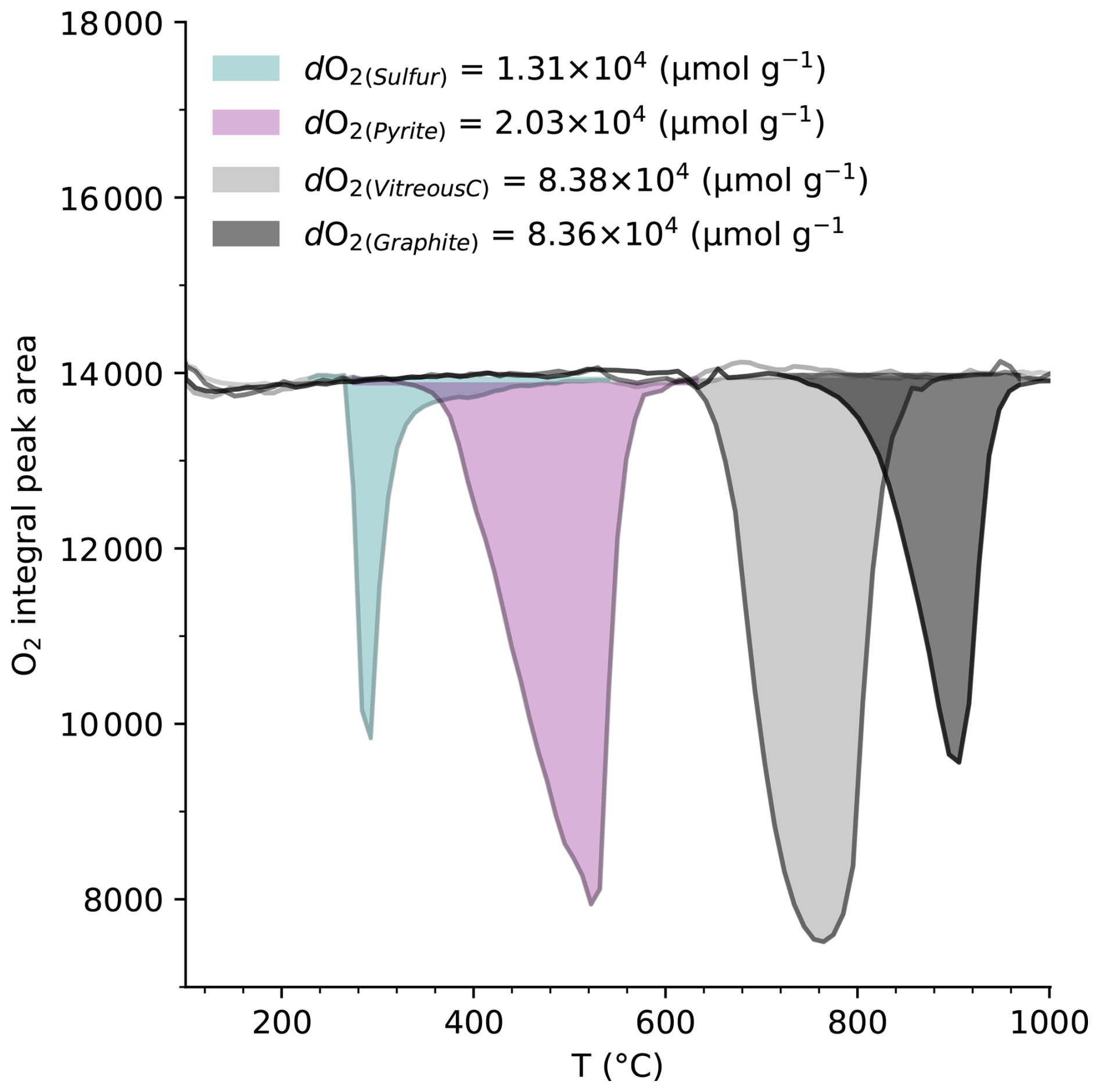
Figure 6Oxygen consumption profiles of elemental sulfur, pyrite, vitreous carbon and graphite during controlled thermal oxidation. Replicate measurements for vitreous carbon and graphite yield high precision and reproductivity (Fig. S1).
The oxidation of various standards was examined to validate this new time-resolved dO2 protocol. The measured dO2 value for pyrite (dO µmol g−1) is approximately 11 % lower than its theoretical values (3.13×104 and 2.29×104 µmol g−1). In contrast, elemental sulfur a dO µmol g−1 corresponding to only ∼42 % of its theoretical value (3.12×104 µmol g−1). This substantial under-recovery indicated that low-temperature volatilization can remove reactive sulfur tom the system before complete oxidation. By contrast, vitreous carbon (dO µmol g−1) and natural graphite (8.36×104 µmol g−1) display excellent alignment with the theoretical prediction (8.36×104 µmol g−1). Their oxidation occurs at higher temperatures (>400° C), where losses due to non-oxidative volatilization are minimal, confirming precision and accuracy of the method for refractory phases. By tracking O2 consumption throughout controlled heating, the method captures the cumulative amount of reduced material that oxidized under imposed analytical conditions (Fig. 6). The integrated O2 consumption across the full temperature range thus provides a quantitative measure of intrinsic redox capacity, while the temperature-resolved profile reveals hoe this capacity is distributed among reactive pools with distinct thermal behavior.
3.4 Secondary oven and optimization of complete oxidation
To address the incomplete oxidation of volatile S and light hydrocarbon species, we introduced a secondary oven downstream of the primary heating chamber (Fig. 1). The oven is held at a constant, adjustable temperature (300–800 °C), allowing volatile intermediates (e.g., S vapor, reduced hydrocarbons) released at °C to remain in the gas stream long enough to undergo near-complete oxidation (Fig. 7A). This configuration reduces losses caused by low-temperature volatilization and improves recovery of reactive volatile species independently of their release temperature in the primary furnace. Measurements on standards including elemental S and pyrite confirm that the secondary oven significantly improve the oxidation efficiency (Fig. 7B, C). For instance, the measured dO2 of pyrite increases from 2.03×104 µmol g−1 (without secondary oven) to 2.28×104 µmol g−1 when the secondary oven was maintained at 800 °C, corresponding to 95 %–99 % of theoretical oxidation capacity (Fig. 7C). Similarly, elemental S and other S-bearing standards displayed consistent increases in dO2 with rising secondary oven temperatures (Fig. 7B). Natural samples from the black shales, Congo Basin peat samples and Lake Cadagno sediments also showed systematic dO2 increases of 10 %–20 % with a secondary oven held at 800 °C (Fig. 7D–F). These results highlight the important role of a secondary furnace in minimizing the low-temperature volatilization losses, particularly for labile sulfur- and fresh organic-rich materials. The secondary oven improves both the accuracy and reproducibility of redox capacity measurements by ensuring more complete oxidation of volatile reaction intermediates.
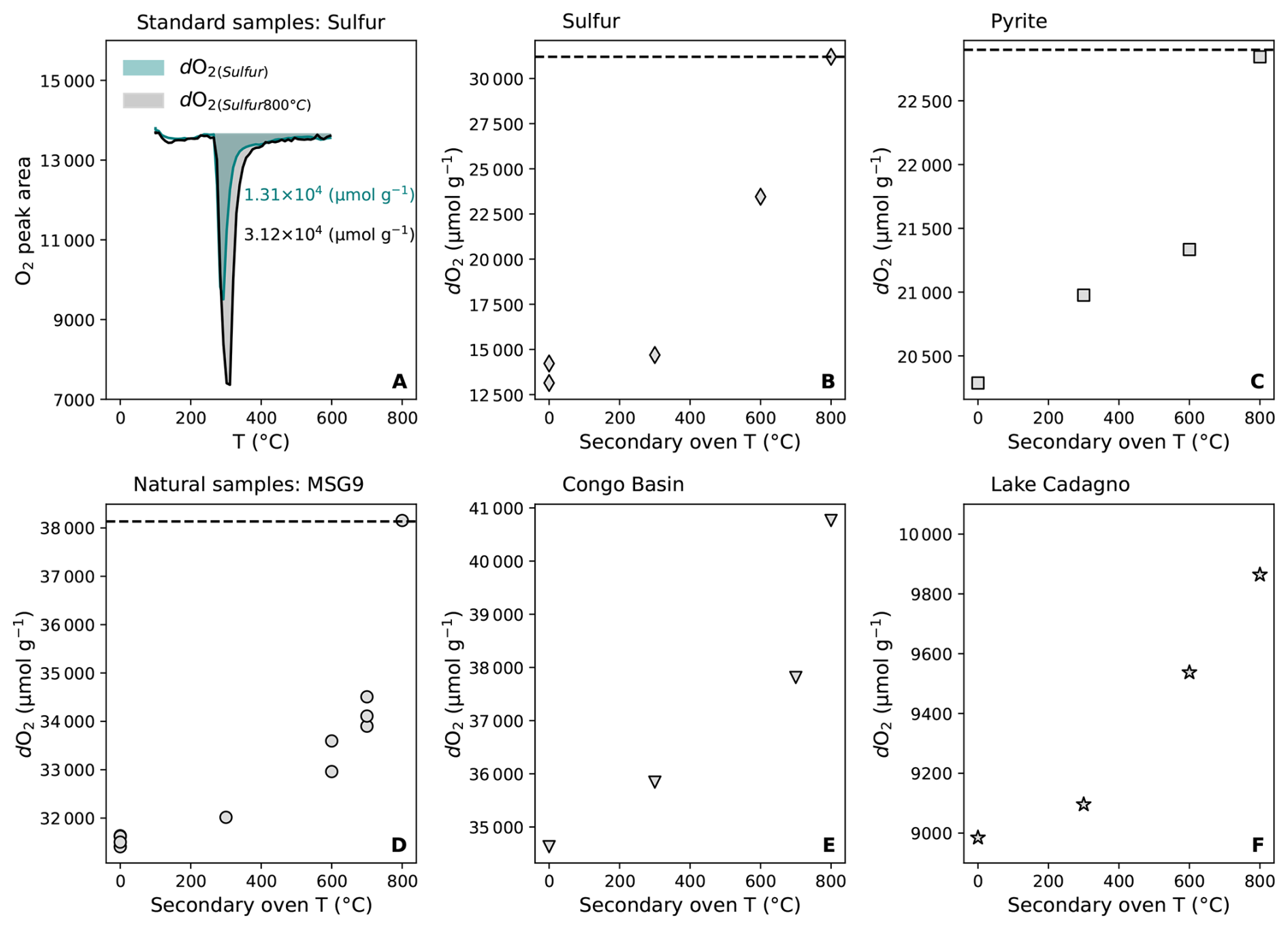
Figure 7Optimized redox capacity with secondary oven. (A), Comparison of elemental sulfur measurements with secondary oven (at 800 °C, grey) and without secondary oven (teal); (B), elemental sulfur; (C), pyrite; (D), black shale (MSG9); (E), peat sediment from the Congo basin; (F), Lake Cadagno sediment. Black dashed lines in (B), (C) and (D) panels indicate the theoretical values.
4.1 Oxygen consumption and redox-active components
Beyond quantifying total redox capacity, our approach yields kinetically resolved oxygen consumption profiles, providing a distinctive redox fingerprint of natural materials. The Congo Basin sample exhibits a bimodal oxidation pattern: ∼37 % of the total O2 corresponds to a low-temperature fraction oxidized in the 250–450 °C window, consistent with labile organic matter, whereas the dominant fraction (63.1 %) is associated with a higher-temperature fraction oxidized about 500 °C, consistent with more refractory organic matter (Fig. 8A). These two oxidation stages are supported with broad CO2 peaks observed in the gas evolution data (Fig. 8D).
The Lake Cadagno sediment exhibits a mixed organic C–S oxidation signature, with 38.0 % of total O2 uptake associated with low-temperature C and S-bearing compounds, and 62.0 % associated with higher-temperature oxidation of refractory carbon and pyrite oxidation extending to ∼700 °C (Fig. 8B). The coupled evolution of CO2, SO2, COS and H2S (Fig. 8E) reflects the co-occurrence of sulfurized organic matter and pyrite, consistent with microbially mediated sulfurization of buried organic matter and pyrite formation under euxinic conditions.
In contrast, the fayalite standard is dominated by Fe(II) oxidation, which accounts for 88.1 % of the total O2 consumption at high temperatures (500–900 °C), with only 11.9 % attributed to minor low-temperature organic matter oxidation (Fig. 8C). This interpretation is consistent with a weak low-temperature CO2 peak and illustrates the method's ability to distinguish metal-driven versus organic- and sulfur driven redox carriers (Fig. 8C, F).
Together, these results demonstrate that integrating O2 consumption profile with simultaneous gas evolution measurements enables the semi-quantitative partitioning of oxygen demand among labile organic matter, refractory carbon, sulfur-bearing phases, and Fe(II) minerals. This provides a kinetically resolved redox characterization of sediments and minerals that cannot be obtained from bulk elemental composition alone.
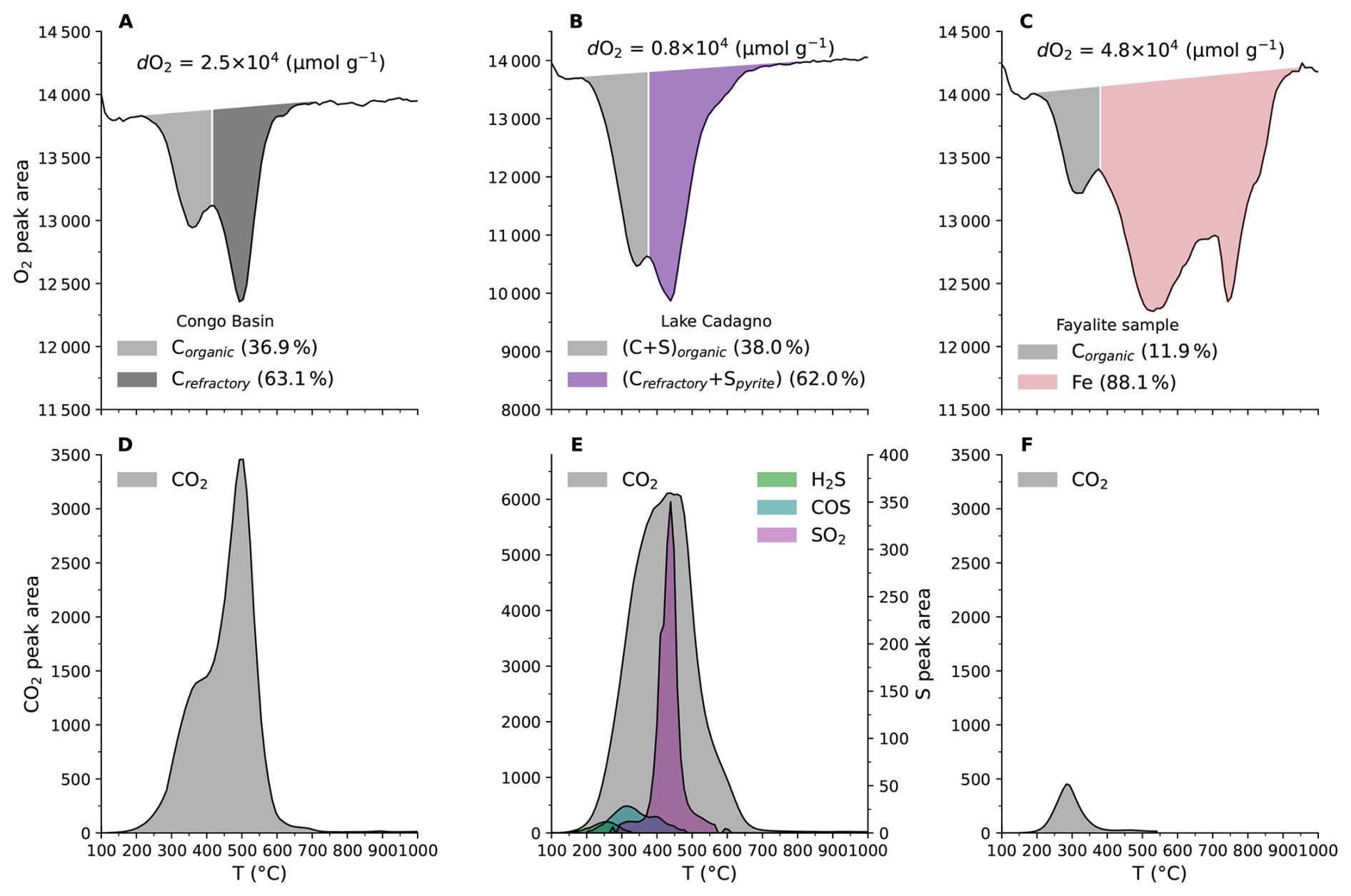
Figure 8Oxidation profile with corresponding components. (A), Congo Basin peat sample; (B), Cadagno Lake sediment sample; (C), fayalite sample; (D–F), gases released through distinct reactions from samples (A), (B) and (C), respectively.
4.2 Sulfur cycle and diagenesis in Lake Cadagno
To further demonstrate the capabilities of our method, we applied it to a sediment profile from Lake Cadagno, a well-known system for studying microbial sulfur cycling and diagenetic processes (Berg et al., 2022; Berg et al., 2025; Dupeyron et al., 2025).
Our results reveal a distinct redox transition marked by a progressive decline in dO2 and total organic carbon (TOC) with depth, while COS and H2S signals persist throughout the profile (Fig. 9A–D). In contrast, SO2 release during analysis are low near the surface, increases sharply to a maximum around 18–20 cm, and then declines toward to the base of the core (Fig. 9E). This hump-shaped SO2 pattern suggests a zone of intense S remobilization and re-oxidation, coinciding with the microbial redox transition zone in which sulfate reduction and sulfide mineral formation were particularly active.
The aEa spectra, derived from the SO2 release kinetics, provide a deeper view into the progression of S diagenesis. In the upper zone (7–15 cm), the spectra exhibit multiple peaks spanning 80–240 kJ mol−1, consistent with metastable metal sulfides, including labile and metastable reduced-S phases, poorly crystalline Fe monosulfides and trace metal sulfides (Fig. 9F). In the transitional middle zone (15–19 cm), the spectra simplify to three partially overlapping peaks, indicating progressive loss of the most labile sulfide phases and their partial transformation into more stable S-bearing minerals (Fig. 9G). Below the 19–20 cm boundary, the spectra converge into a single dominant peak at approximately 140–160 kJ mol−1, consistent with the predominance of more thermally stable pyrite (Fig. 9H). This clear diagenetic transition from diverse assemblage of reactive sulfides to a more uniform and thermally stable phase of pyrite highlights the multi-stage and progressive nature of sulfur mineral maturation in anoxic sediments.
We interpret the 19–20 cm boundary as marking a sulfate-depletion front, consistent with recent study (Dupeyron et al., 2025). The transformation into stable pyrite is consistent with a reduction in the pool of reactive sulfur species and progressive sulfur mineral maturation with depth. By resolving activation-energy distributions, our method provides a new kinetic framework for tracking diagenetic fronts and reconstructing microbial sulfur cycling and redox transitions in anoxic environments.
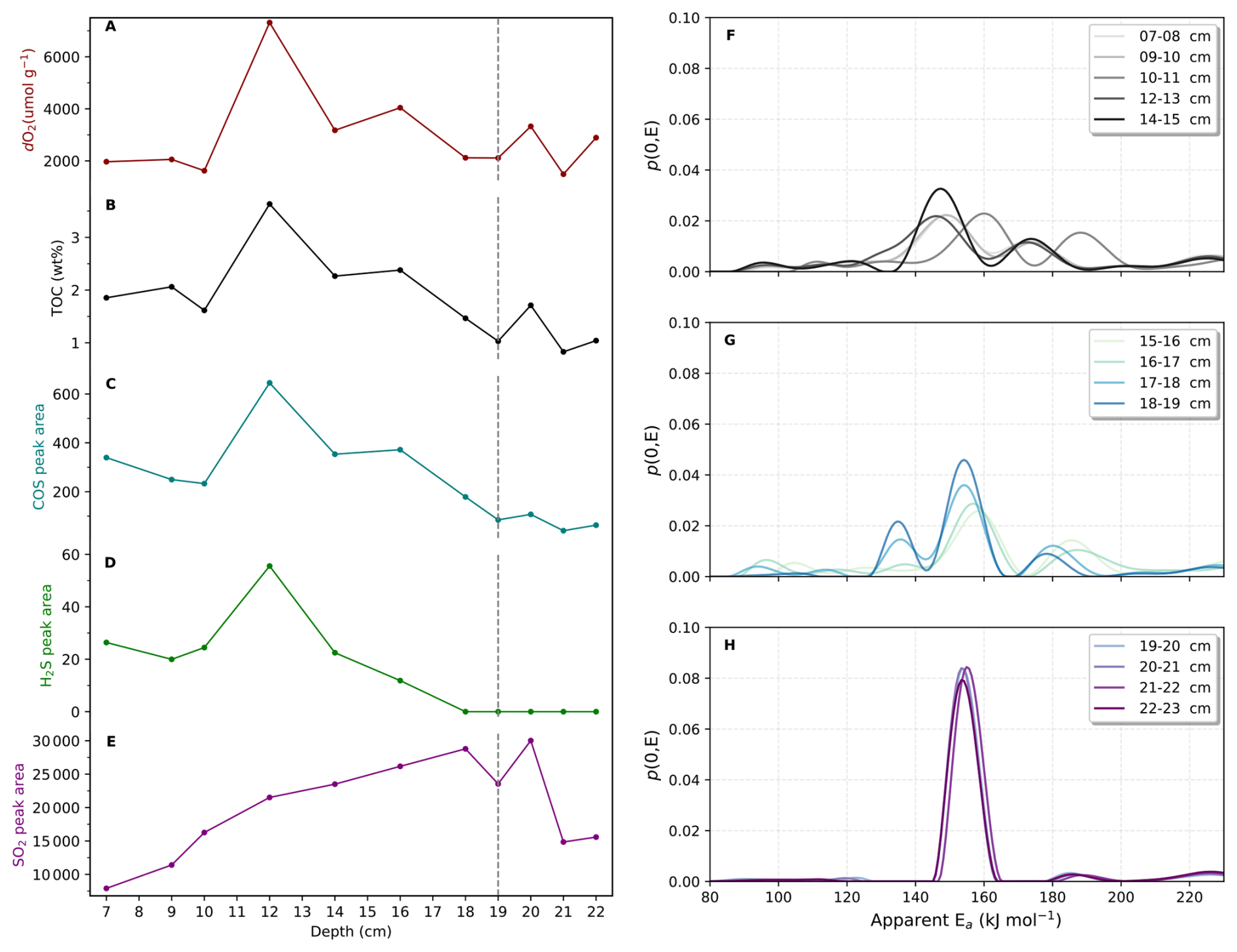
Figure 9Cadagno Lake depth profile. (A), dO2; (B), TOC content; (C), COS; (D), H2S; (E), SO2; (F), aEa spectral distributions of SO2 from the upper zone (7–15 cm); (G), aEa spectral distributions of SO2 from the middle zone (15–19 cm). (H), aEa spectral distributions of SO2 from the lower zone (19–22 cm). Dash line indicates the diagenetic boundary.
4.3 Kinetic fingerprint of materials: redox chemistry
The TGA/DSC-MicroGC platform delivers a comprehensive combustion kinetic fingerprint, that is, a spectral signature of H, C, O, and S reactivity, captured in a single analytical run under controlled ramped oxidation. Fig. 10 represents the kinetic fingerprint for a representative sulfidic sediment from Lake Cadagno. It shows the progressive sample mass loss and the corresponding heat-flow profile, which together reflect oxidation reactions and associated bond transformations (Fig. 10A, B). Moreover, it tracks the oxidation-consumption profile and the evolution of diagnostic gases, including C- and S-bearing species (CO2, COS, SO2 and H2S) (Fig. 10C, D and E). The apparent activation energy distributions further resolve the thermal reactivity of these phases. Low aEa (∼ 100–140 kJ mol−1) associated with H2S and COS evolution, are consistent with thermally labile organic S-bearing compounds, including sulfurized organic matter and/or, metastable sulfide phases as well as more labile carbon pools carbon. In contrast high aEa (∼ 150–180 kJ mol−1) associated with SO2 and CO2 evolution are consistent with the oxidation of more refractory phases, including pyrite and thermally stable carbon (Fig. 10F).
By synchronously resolving mass-loss kinetics, enthalpic transitions, activation-energy distributions, and near-continuous evolved gas profiles (Fig. 10), this approach provides a kinetically resolved characterization of redox-active phases. It links thermal stability, gas evolution and oxygen demand within a unified analytical framework.
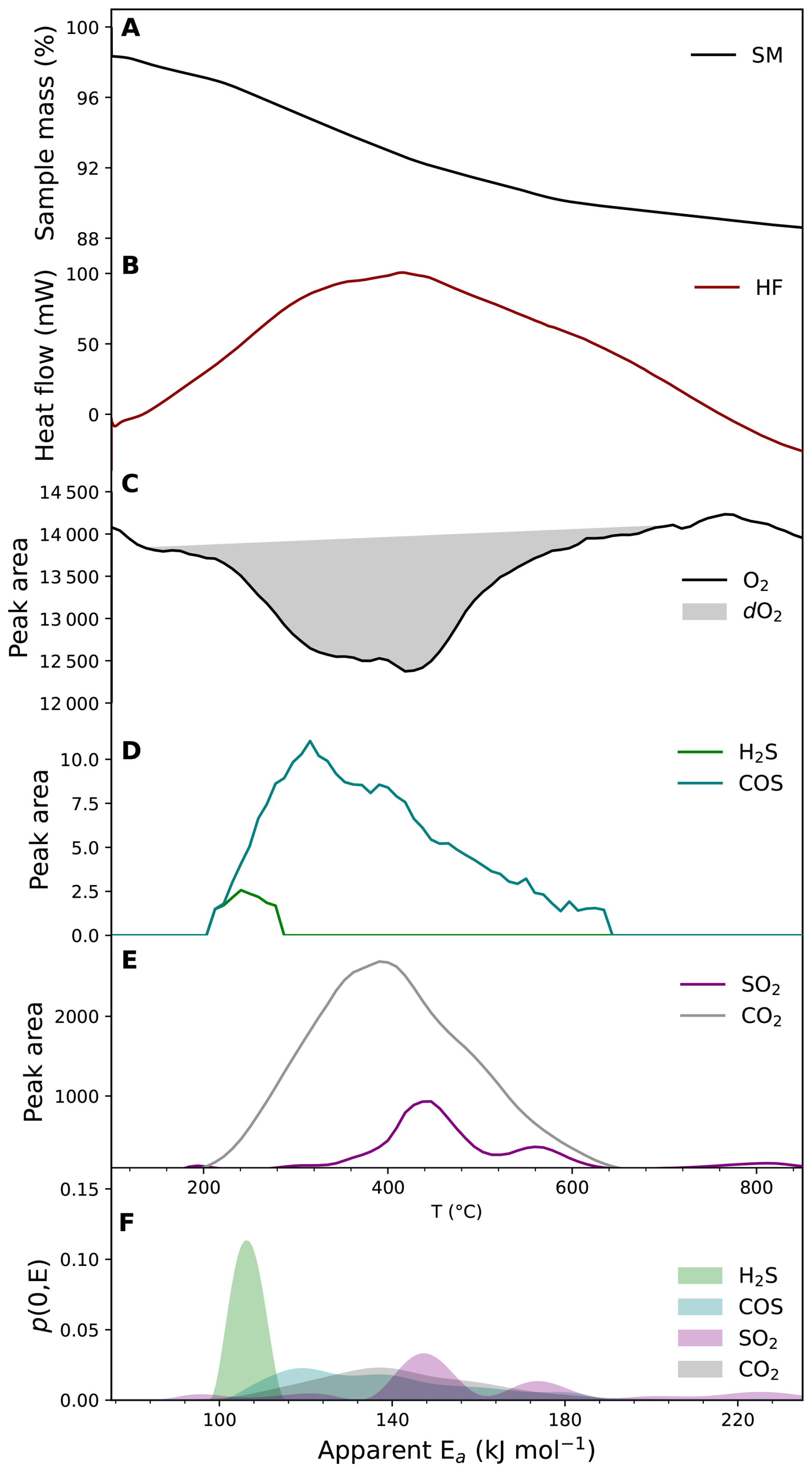
Figure 10Kinetic fingerprint for a representative sulfidic sediment from Lake Cadagno. (A), Sample mass (SM) changes at heating rates of 10 °C min−1; (B), Heat flow (HF) profile; (C), oxidation profile; (D), H2S and COS profiles; (E), SO2 and CO2 profiles; (F), aEa distribution of H2S, COS, SO2 and CO2.
In this study, we present a novel, integrated TGA/DSC-MicroGC analytical system that enables kinetically resolved characterization of geological materials using high-resolution thermal analysis. By combining thermogravimetric analysis, differential scanning calorimetry and ultra-fast gas chromatography within a single experimental framework, this method provides a mechanistic approach for distinguishing complex C- and S-bearing reactive pools based on their thermal reactivity and gas-evolution signature It also quantifies total and fractional redox capacity (oxidability) through time-resolved monitoring of oxygen demand during controlled ramped oxidation. These insights about elemental speciation, reaction kinetics, and redox signature cannot be resolved using conventional bulk analyses alone.
Applications to natural archives from the Congo Basin and Lake Cadagno demonstrate the method's broad utility and environmental relevance. In Congo Basin peatland sediments, the method differentiated thermally labile and refractory organic carbon pools and quantifies their contrasting oxygen demand, providing a new framework for tracing changes in organic matter preservation and paleoenvionmental redox dynamics (Galvez et al., 2026). In Lake Cadagno sediments, our results reveal a diagenetic front defined by a shift in sulfide speciation and apparent activation-energy, providing a direct kinetic proxy for past microbial S cycling and redox evolution. More broadly, the integration of oxygen consumption profiles with evolved gas signatures allows semi-quantitative partitioning of redox-active components among organic matter, sulfur-bearing phases, and Fe-bearing minerals, yielding a kinetic “redox fingerprint” of geological materials.
Overall, this integrated approach moves beyond static compositional measurements toward a mechanistic, high-resolution framework for tracing coupled C-O-S dynamics in Earth materials. The TGA/DSC-MicroGC platform provides a versatile tool for investigating redox conditions, paleoenvironmental change, and biogeochemical reactivity across diverse natural systems. Future applications to marine sediments, soils, and ancient rock archives may further refine our understanding of redox processes across environmental settings and their links to elemental cycling and Earth evolution.
The analysis scripts generated for this study are highly customized to the specific data infrastructure and require specific explanation for use. The code is, however, available upon reasonable request from the corresponding author, who can provide the necessary support and documentation. All relevant data related to this study are available via Zenodo (https://doi.org/10.5281/zenodo.20739985).
The supplement related to this article is available online at https://doi.org/10.5194/bg-23-4859-2026-supplement.
Conceptualization: M.E.G.; Investigation: S.W., M.E.G.; Methodology: S.W., M.E.G. Writing – original draft: S.W.; Writing – review and editing: S.W., S.L.J., M.E.G.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
This project was supported through Swiss National Science Foundation grant 215097 and a Branco Weiss Society in Science fellowship (M.E.G.). This study was also supported by the Chinese Academy of Sciences-Pioneer Hundred Talents Program (grant E5710403 awarded to S. W) and the Swiss National Science Foundation (grants 200021_163003 and 200020_192361 awarded to S.L.J.). We thank J. Dupeyron, J. Marin Carbonne and Y. Garcin for proving the sediment samples from the Lake Cadagno and the Congo Basin as well as for their helpful discussions.
This research has been supported through Swiss National Science Foundation (grant 215097, grants 200021_163003 and 200020_192361) and the Chinese Academy of Sciences-Pioneer Hundred Talents Program (grant E5710403).
This paper was edited by Sebastian Naeher and reviewed by Małgorzata Labus and one anonymous referee.
Alt, J. C. and Shanks III, W. C.: Stable isotope compositions of serpentinite seamounts in the Mariana forearc: Serpentinization processes, fluid sources and sulfur metasomatism, Earth Planet Sci. Lett., 242, 272–285, https://doi.org/10.1016/j.epsl.2005.11.063, 2006.
Berg, J. S., Lepine, M., Laymand, E., Han, X., Vogel, H., Morlock, M. A., Gajendra, N., Gilli, A., Bernasconi, S. M., Schubert, C. J., Su, G., and Lever, M. A.: Ancient and Modern Geochemical Signatures in the 13,500-Year Sedimentary Record of Lake Cadagno, Front Earth Sc.-Switz., 9, https://doi.org/10.3389/feart.2021.754888, 2022.
Berg, J. S., Rodriguez, P. C., Magnabosco, C., Deng, L., Bernasconi, S. M., Vogel, H., Morlock, M., and Lever, M. A.: Microbial sulfur cycling across a 13 500-year-old lake sediment record, Biogeosciences, 22, 5483–5496, https://doi.org/10.5194/bg-22-5483-2025, 2025.
Boudreau, B. P.: A kinetic model for microbic organic-matter decomposition in marine sediments, FEMS Microbiol. Ecol., 11, 1–14, https://doi.org/10.1111/j.1574-6968.1992.tb05789.x, 1992.
Carter, T. L., Schaecher, C., Monteith, S., and Ferguson, R.: Using combustion analysis to simultaneously measure soil organic and inorganic carbon, Geoderma, 451, https://doi.org/10.1016/j.geoderma.2024.117066, 2024.
Cohen-Sadon, H., Amrani, A., Feinstein, S., and Rosenberg, Y. O.: A new empirical approach for rapid quantification of organic and pyritic sulfur in sedimentary rocks using the Rock-Eval 7S, Org. Geochem., 166, 104350, https://doi.org/10.1016/j.orggeochem.2021.104350, 2022.
Crezee, B., Dargie, G. C., Ewango, C. E. N., Mitchard, E. T. A., Emba B, O., Kanyama T, J., Bola, P., Ndjango, J.-B. N., Girkin, N. T., Bocko, Y. E., Ifo, S. A., Hubau, W., Seidensticker, D., Batumike, R., Imani, G., Cuní-Sanchez, A., Kiahtipes, C. A., Lebamba, J., Wotzka, H.-P., Bean, H., Baker, T. R., Baird, A. J., Boom, A., Morris, P. J., Page, S. E., Lawson, I. T., and Lewis, S. L.: Mapping peat thickness and carbon stocks of the central Congo Basin using field data, Nat. Geosci., 15, 639–644, https://doi.org/10.1038/s41561-022-00966-7, 2022.
Dupeyron, J., Pasquier, V., Guibourdenche, L., Busigny, V., Cartigny, P., Jézéquel, D., Bernasconi, S. M., and Carbonne, J. M.: Decadal, sediment-driven sulfur isotope evolution in Lake Cadagno, Geochem. Perspect. Lett., 38, 17–22, https://doi.org/10.7185/geochemlet.2550, 2025.
Espitalie, J., Deroo, G., and Marquis, F.: Rock-Eval Pyrolysis and Its Applications (Part Two), Rev. Inst. Fr. Pét., 40, 755–784, https://doi.org/10.2516/ogst:1985045, 1985.
Galvez, M., Wu, S., Garcin, Y., Schefuß, E., Gassier, G., Lebamba, J., Kiahtipes, C., Bokomba, F., Wotzka, H. P., and Adatte, T.: Hydroclimate controls on Congo peatland net oxygen release over the past 10,600 years, Nat. Geosci., https://doi.org/10.1038/s41561-026-02031-z, 2026.
Galvez, M. E.: Redox constraints on a Cenozoic imbalance in the organic carbon cycle, Am. J. Sci., 320, 730–751, https://doi.org/10.2475/10.2020.03, 2020.
Galvez, M. E. and Jaccard, S. L.: Redox capacity of rocks and sediments by high temperature chalcometric titration, Chem. Geol., 564, 120016, https://doi.org/10.1016/j.chemgeo.2020.120016, 2021.
Galvez, M. E., Fischer, W. W., Jaccard, S. L., and Eglinton, T. I.: Materials and pathways of the organic carbon cycle through time, Nat. Geosci., 13, 535–546, https://doi.org/10.1038/s41561-020-0563-8, 2020.
Galvez, M. E., Müntener, O. and Jaccard, S. L.: Beyond Oxygen Fugacity: A Compositional Metric to Probe Earth's Redox Structure, Geophys. Res. Lett., 52, https://doi.org/10.1029/2025GL117642, 2025.
Garcin, Y., Schefuß, E., Dargie, G. C., Hawthorne, D., Lawson, I. T., Sebag, D., Biddulph, G. E., Crezee, B., Bocko, Y. E., Ifo, S. A., Mampouya Wenina, Y. E., Mbemba, M., Ewango, C. E. N., Emba, O., Bola, P., Kanyama Tabu, J., Tyrrell, G., Young, D. M., Gassier, G., Girkin, N. T., Vane, C. H., Adatte, T., Baird, A. J., Boom, A., Gulliver, P., Morris, P. J., Page, S. E., Sjögersten, S., and Lewis, S. L.: Hydroclimatic vulnerability of peat carbon in the central Congo Basin, Nature, 612, 277–282, https://doi.org/10.1038/s41586-022-05389-3, 2022.
Hayes, J. M. and Waldbauer, J. R.: The carbon cycle and associated redox processes through time, Philos. T. Roy. Soc. B, 361, 931–950, https://doi.org/10.1098/rstb.2006.1840, 2006.
Hemingway, J. D., Rothman, D. H., Rosengard, S. Z., and Galy, V. V.: Technical note: An inverse method to relate organic carbon reactivity to isotope composition from serial oxidation, Biogeosciences, 14, 5099–5114, https://doi.org/10.5194/bg-14-5099-2017, 2017.
Hemingway, J. D., Hilton, R. G., Hovius, N., Eglinton, T. I., Haghipour, N., Wacker, L., Chen, M.-C., and Galy, V. V.: Microbial oxidation of lithospheric organic carbon in rapidly eroding tropical mountain soils, Science, 360, 209–212, https://doi.org/10.1126/science.aao6463, 2018.
Janssen, D. J., Rickli, J., Wille, M., Sepúlveda Steiner, O., Vogel, H., Dellwig, O., Berg, J. S., Bouffard, D., Lever, M. A., Hassler, C. S., and Jaccard, S. L.: Chromium Cycling in Redox-Stratified Basins Challenges δ 53Cr Paleoredox Proxy Applications, Geophys. Res. Lett., 49, https://doi.org/10.1029/2022GL099154, 2022.
L'Vov, B. V.: Mechanism of thermal decomposition of alkaline-earth carbonates, Thermochim. Acta, 303, 161–170, https://doi.org/10.1016/S0040-6031(97)00261-X, 1997.
Ordoñez, L., Vogel, H., Sebag, D., Ariztegui, D., Adatte, T., Russell, J. M., Kallmeyer, J., Vuillemin, A., Friese, A., Crowe, S. A., Bauer, K. W., Simister, R., Henny, C., Nomosatryo, S., and Bijaksana, S.: Empowering conventional Rock-Eval pyrolysis for organic matter characterization of the siderite-rich sediments of Lake Towuti (Indonesia) using End-Member Analysis, Org. Geochem., 134, 32–44, https://doi.org/10.1016/j.orggeochem.2019.05.002, 2019.
Paytan, A., Kastner, M., Campbell, D., and Thiemens, M. H.: Sulfur Isotopic Composition of Cenozoic Seawater Sulfate, Science, 282, 1459–1462, https://doi.org/10.1126/science.282.5393.1459, 1998.
Salonen, K.: A versatile method for the rapid and accurate determination ofcarbon by high temperature combustion, Limnol. Oceanogr., 24, 177–183, https://doi.org/10.4319/lo.1979.24.1.0177, 1979.
Sebag, D., Verrecchia, E. P., Cécillon, L., Adatte, T., Albrecht, R., Aubert, M., Bureau, F., Cailleau, G., Copard, Y., Decaens, T., Disnar, J. R., Hetényi, M., Nyilas, T., and Trombino, L.: Dynamics of soil organic matter based on new Rock-Eval indices, Geoderma, 284, 185–203, https://doi.org/10.1016/j.geoderma.2016.08.025, 2016.
Vyazovkin, S. and Wight, C. A.: Model-free and model-fitting approaches to kinetic analysis of isothermal and nonisothermal data, Thermochim. Acta, 340-341, 53–68, https://doi.org/10.1016/S0040-6031(99)00253-1, 1999.
Yoon, G., Park, S.-M., Yang, H., Tsang, D. C. W., Alessi, D. S., and Baek, K.: Selection criteria for oxidation method in total organic carbon measurement, Chemosphere, 199, 453–458, https://doi.org/10.1016/j.chemosphere.2018.02.074, 2018.
Zhao, W. Z., Lu, B., Lv, S. N., Zhou, C. F., and Yang, Y.: Simultaneous determination of chlorine and sulfur in geochemical reference samples by wavelength dispersive X-ray fluorescence spectrometry, New J. Chem., 44, 11224–11230, https://doi.org/10.1039/d0nj02042g, 2020.
We present a novel analytical system that simultaneously measures mass loss, heat flow, gas emissions, and oxygen consumption during controlled heating. The method reveals how carbon, oxygen, and sulfur are stored, transformed, and recycled in geological materials. By providing high-resolution insights into thermal reactivity and redox processes, it helps identify hidden environmental changes and offers a powerful new tool for studying biogeochemical cycles and Earth’s environmental evolution.
We present a novel analytical system that simultaneously measures mass loss, heat flow, gas...